
MOOC Estimation des incertitudes de mesure en analyse chimique
10. Aperçu des approches d'estimation de l'incertitude de mesure
10.5. Incertitude-type des données d'entrée
https://sisu.ut.ee/measurement/95-step-5-%E2%80%93-standard-uncertainties-input-quantities
Incertitude type des données d'entrée
http://www.uttv.ee/naita?id=17641
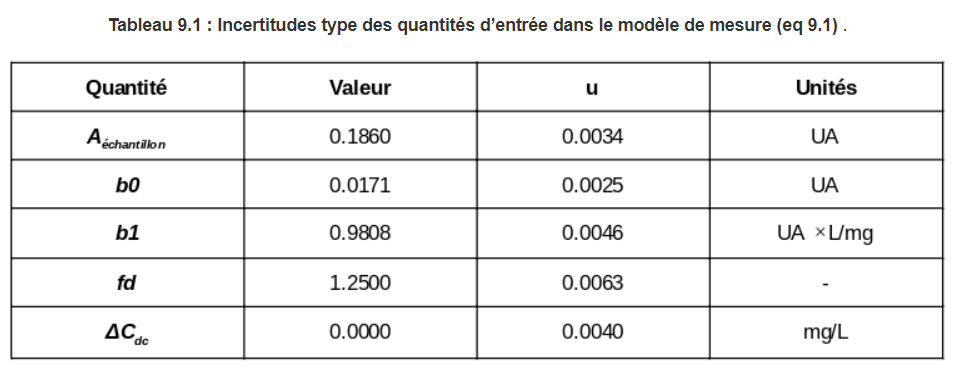
| The next step is finding standard uncertainties of the input quantities. We will now look at all the input quantities that we had in our model one by one. And the first input quantity is absorbance of the sample solution. As we saw previously when we looked at uncertainty sources, there are three main uncertainty sources to this input quantity : repeatability of the photometric reading, possible drift of the photometer properties and also possible chemical interferences which can come either by increased absorbance because some other compounds also absorb at used wavelength or by decreased absorbance because something causes the dye either not to form or to a little bit partially decompose. And here the standard uncertainties corresponding to those uncertainty sources are presented. The repeatability standard uncertainty can be easily determined in the laboratory just by measuring repeatedly absorbance of the same solution over a few hours of time. But of course, each time the spectrophotometer cell with the solution should be inserted again and then removed. Now the drift uncertainty can likewise be determined in the laboratory but also it sometimes can be found in the documentation of the spectrophotometer. And what can be said here is that these values correspond to quite safe estimates for contemporary normal usual routine spectrophotometer. Now this uncertainty due to the chemical interferences is more difficult to estimate and actually this estimate can only come by experience from the laboratory unless extensive scientific research is undertaken. In the case of ammonium determination, in most samples we have found that this uncertainty estimate 0.003 absorbance units is fairly useful for the concentration range that is currently under investigation. And now since all these uncertainty components are related to the same quantity, expressed in the units of that same quantity and also expressed as standard uncertainties, we can simply combine them using the squared summing rule which we have seen already previously. We square all the components, sum them up, take square root and find that the combined standard uncertainty of absorbance of the sample solution is 0.0034 absorbance units. The next input quantities are the intercept and slope of the calibration line : b0 and b1. And here we take an approximate solution or an approximate approach. We estimate the standard uncertainties of these two input quantities as simple standard deviations of those quantities as found by regression data analysis, so that their standard uncertainties are given here. And now what does this approximation mean ? It means two things : on one hand it takes into account only the random effects that are evident in these quantities, meaning possible systematic effects will be rejected. However usually such systematic effects and calibration graphs, if they are correctly made, are quite small. Nevertheless, we slightly underestimated uncertainty. On the other hand, all the equations that we are looking at and in particular the equation that comes at the end for calculating the combined standard uncertainty of our measurand they assume non-correlated input quantities. Now, slope and intercept of the same calibration line are always slightly correlated but this correlation is a negative correlation meaning if one of them becomes higher then the other one becomes lower and if a negative correlation is left out of consideration then we slightly overestimate the uncertainty. So, we on one hand somewhat underestimate measurement uncertainty. On the other hand we somewhat overestimate measurement uncertainty. These two effects largely cancel resulting in a very realistic uncertainty estimate. The next input quantity is the dilution factor and dilution factor is nothing else than ratio of different volumes. In our case 50 to 40 ratio resulting in the dilution factor value 1.25. And in this case we will not look in detail at all the volumetric operations because as we will discover later on, the volumetric uncertainty in this uncertainty batch is not very significant. Instead we take a rather safe and very effective approach. We estimate the standard uncertainty of dilution factor as 0.5% of the dilution factor value. As we found according to the experience from our laboratory, this is almost always very suitable uncertainty estimate if volumetric operations are performed correctly. So that the dilution factor standard uncertainty will be 0.5% from 1.25 meaning 0.0063 and it is a unitless quantity. The next input quantity is the ΔCdc. As we saw, this input quantity is meant for taking into account possible decomposition of ammonium or possible contamination of the sample of the solution. I stress here the word possible because it can well be that there's neither contamination nor decomposition but since we do not exactly know, we have to take this possible effect into account as an uncertainty. And this uncertainty unless serious scientific research is carried out, cannot be easily rigorously estimate. So that again we have to use a value which is based on long-term experience. And at this concentration level, it has been found that 0.004 mg/L as standard uncertainty is a very suitable estimate for normal usual conditions of ammonium determination. Now we have found the standard uncertainties of all the input quantities and this slide demonstrates the summary of this. We now have all values and all standard uncertainties and we now can proceed with uncertainty estimation. | L'étape suivante consiste à trouver les incertitudes types des données d'entrée. Nous allons maintenant aborder l'ensemble des données d'entrée que nous avions dans notre modèle une à une. Et la première donnée d'entrée est l'absorbance de la solution d'échantillon. Comme nous l'avons vu précédemment lorsque nous avons abordé les sources d'incertitude, il en existe trois principales qui sont liées à cette donnée d'entrée : la répétabilité de la lecture photométrique, l'éventuelle dérive des propriétés du photomètre et aussi d'éventuelles interférences chimiques qui peuvent provenir soit d'une augmentation de l'absorbance causée par d'autres composés qui absorbent également à la longueur d'onde utilisée ou soit d'une diminution de l'absorbance causée par quelque chose qui empêche la teinte de se former ou qui entraîne en partie sa décomposition. Les incertitudes types qui correspondent à ces sources d'incertitude sont présentées ici. L'incertitude type de répétabilité peut être facilement déterminée au sein du laboratoire en mesurant à plusieurs reprises l'absorbance d'une même solution sur une période de quelques heures. La cellule du spectrophotomètre qui contient la solution doit évidemment être insérée puis retirée lors de chaque mesure. On peut également déterminer l'incertitude de dérive au sein du laboratoire mais on peut aussi parfois la trouver dans la documentation du spectrophotomètre. Ce que l'on peut dire ici c'est que ces valeurs correspondent à des estimations assez certaines pour un spectrophotomètre de routine habituel et récent. L'incertitude causée par les interférences chimiques est plus difficile à estimer et en réalité cette estimation ne peut être trouvée que grâce à l'expérience acquise au sein du laboratoire à moins que des recherches scientifiques approfondies ne soient entreprises. Dans le cas de la détermination de l'ammonium, dans la plupart des échantillons nous avons constaté que cette estimation de l'incertitude qui est à 0,003 unités d'absorbance est assez utile pour la gamme de concentration qui est actuellement étudiée. Puisque toutes les composantes de l'incertitude sont à la fois liées à la même donnée, exprimées en unités de cette même donnée et également exprimées comme incertitudes types, nous pouvons simplement les associer en utilisant la règle de sommation au carré que nous avons vu précédemment. On élève au carré tous les composants, on les additionne, on prend la racine carrée et on trouve que l'incertitude-type composée de l'absorbance de la solution d'échantillon est de 0,0034 unités d'absorbance. Les autres données d'entrées sont l'ordonnée à l'origine et la pente de la droite d'étalonnage: b0 et b1. Ici on prend une solution approximative ou on se place dans une approche approximative. On estime que les incertitudes types de ces deux données d'entrée sont de simples écarts-types de ces données, trouvés par l'analyse des données de régression, de sorte que leurs incertitudes types sont données ici. Que signifie cette approximation? Cela signifie deux choses : d'une part elle ne prend en compte que les effets aléatoires qui sont évidents dans ces données, ainsi les éventuels effets systématiques seront négligés. Cependant, les effets systématiques sont généralement assez faibles si les graphiques d'étalonnage sont correctement réalisés. Néanmoins, nous avons légèrement sous-estimé l'incertitude. D'autre part, les équations que nous examinons et en particulier l'équation qui vient à la fin pour calculer l'incertitude type composéede notre mesurande supposent toutes des données d'entrée non corrélées. La pente et l'ordonnée à l'origine de la même droite d'étalonnage sont toujours légèrement corrélés mais cette corrélation est négative, cela signifie que si l'une augmente alors l'autre diminue et si l'on ne tient pas compte d'une corrélation négative alors on surestime légèrement l'incertitude. D'une part, on sous-estime ainsi quelque peu l'incertitude de mesure. D'autre part on surestime quelque peu l'incertitude de mesure. Ces deux effets s'annulent en grande partie, on obtient ainsi une estimation très réaliste de l'incertitude. La prochaine donnée d'entrée est le facteur de dilution et ce facteur n'est rien d'autre qu'un rapport de différents volumes. Dans notre cas, il s'agit du rapport de 50 sur 40. On obtient un facteur de dilution de 1,25. Dans ce cas nous ne regarderons pas en détail toutes les opérations volumétriques car, comme nous le verrons plus tard, l'incertitude volumétrique dans ce groupe d'incertitudes n'est pas très significative. Au lieu de cela, nous adoptons une approche plutôt sûre et très efficace. On estime que l' incertitude type du facteur de dilution représente 0,5% de la valeur de ce facteur de dilution. Comme nous l'avons constaté lors de l'expérience réalisée dans notre laboratoire, l'estimation de l'incertitude est presque toujours très convenable si les opérations volumétriques sont effectuées correctement. L'incertitude type du facteur de dilution sera ainsi de 0,5% de 1,25 ce qui correspond à 0,0063 et cette donnée obtenue est sans unité. Une autre donnée d'entrée est le ΔCdc. Comme nous l'avons vu, le rôle de cette donnée d'entrée est de prendre en compte la décomposition possible de l'ammonium ou la contamination possible de l'échantillon de la solution. J'insiste ici sur le mot possible car il se peut bien qu'il n'y ait ni contamination ni décomposition mais comme nous ne le savons pas exactement, nous devons tenir compte de cet effet possible comme d'une incertitude. Cette incertitude ne peut pas être facilement évaluée avec rigueur à moins que des recherches scientifiques sérieuses ne soient effectuées. C'est pour cela que nous devons à nouveau utiliser une valeur basée sur une expérience au long terme. A ce niveau de concentration, on considère qu'une incertitude type de 0,004 mg/L est une estimation très convenable pour les conditions normales normales de détermination de l'ammonium. Maintenant, nous avons trouvé les incertitudes types de toutes les données d'entrée comme le résume cette diapositive. Nous avons maintenant toutes les valeurs et toutes les incertitudes types et nous pouvons dès lors procéder à l'estimation de l'incertitude. |
Trouver les incertitudes types des données d’entrée :
L’incertitude type de l’absorbance de l’échantillon (Aéchantillon) est trouvée à partir des 3 composantes de l’incertitude suivantes :
1. L’incertitude liée à la répétabilité de la mesure photométrique :
u (Aéchantillon, rep) = 0.0010 UA (9.2)
Cette incertitude englobe la répétabilité de l’instrument, la répétabilité du positionnement de la cellule de détection, et les possibles interférences, comme des particules de poussières sur les fenêtres optiques de la cellule.
2. L’incertitude liée à l’éventuelle dérive des paramètres du spectrophotomètre :
u (Aéchantillon, dérive) = 0.0012 UA (9.3)
3. L’incertitude liée aux possibles effets d’interférence :
u (Aéchantillon, chim) = 0.0030 UA (9.4)
Elle peut être causée par d’autres composés (interférents) qui absorberaient (ou diffuseraient) la lumière à la longueur d’onde utilisée pour la mesure (menant à l’augmentation de la valeur d’absorbance) ou à des perturbations dans le complexe photométrique en formation (menant à la diminution de la valeur d’absorbance).
En résulte :

Nous voyons que l’incertitude liée à de possibles interférences domine de part sa contribution à l’incertitude totale de l’absorbance de l’échantillon. Bien que dans cet exemple quantifier d’éventuelles interférences est facile, en réalité l’évaluation quantitative de l’incertitude liée aux possibles interférences est difficile. Quelques conseils pratiques à ce sujet sont donnés plus bas dans la section “Évaluer l’incertitude liée aux possibles interférences”.
Les incertitude types de la pente b1 et de l’ordonnée à l’origine b0 sont retrouvées comme des écarts-types des coefficients de régression respectifs (voir les fichiers excel de la section 9.7). C’est une manière approximative de prendre en compte l'incertitude liée à l’analyse par régression linéaire, car :
- On néglige les effets systématiques qui affectent tous les points de la droite de régression.
- On néglige la corrélation négative entre b1 et b0 (qui existe toujours).
Le premier de ces effets mène à une sous-estimation de l’incertitude et le second à une surestimation de l’incertitude. Donc, cette approche ne devrait être utilisée que si on ne s’attend pas à ce que l’analyse par régression linéaire soit parmi les principaux contributeurs à l’incertitude. [1]
L’incertitude liée au facteur de dilution u (fd) est trouvée en émettant l’hypothèse que l’incertitude-type relative composée de toutes les opérations volumétriques concernées ne dépasse pas 0.5%. Si les opérations volumétriques sont menées consciencieusement, alors cette hypothèse est raisonnable dans des conditions usuelles de manipulations en laboratoire. Si nous considérons que la valeur de fd est de 1.25 (sans unités), nous obtenons :
u (fd) = 1.25 x 0.5% / 100% = 0.0065 (sans unités) (9.6)
L’incertitude de ΔCdc compte pour la possible décomposition du complexe photométrique et la possible contamination de l’échantillon. Le mot “possible” est accentué ici : il est bien possible qu’en fait, il n’y ait ni de décomposition du complexe photométrique, ni de contamination de l’échantillon. Cependant, afin de rigoureusement établir ceci, des recherches extensives seraient nécessaires. Par conséquent dans cet exemple, nous utilisons une estimation basée sur l’expérience du laboratoire :
u (ΔCdc) = 0.004 mg/L
Les incertitudes types des quantités d’entrée sont résumées dans le tableau suivant :
Évaluer l’incertitude liée aux possibles interférences :
Il n’existe pas de manière universellement applicable pour évaluer l’incertitude liée à de possibles interférences. L’approche la plus rigoureuse est de déterminer séparément le contenu des composés interférents dans l’échantillon et de corriger le résultat (alors évidemment l’incertitude de correction doit être évaluée, mais cela est facile). Cependant, dans la plupart des cas, mener des analyses séparées pour déterminer les (possibles) interférents est bien trop coûteux en travail pour être pratique. La chose est d'avantage rendue compliquée par la quantité limitée d’informations généralement disponibles sur les interférents : dans la plupart des cas, on ne sait pas quels sont les composés qui causent l’interférence. Pour cette raison, si l’interférence n’est pas trop importante alors l’approche habituelle est d’essayer de la prendre en compte en augmentant l’incertitude de mesure [2].
Les 2 exemples qui suivent traitent de comment l’incertitude liée à de possibles composés interférents peut être prise en compte dans l’estimation de l’incertitude de mesure.
1- Obtenir et utiliser des informations sur l’interférence à partir du spectre.
Le schéma qui suit présente une situation où l’interférence est clairement présente et peut être observée à partir de l’apparence du spectre :
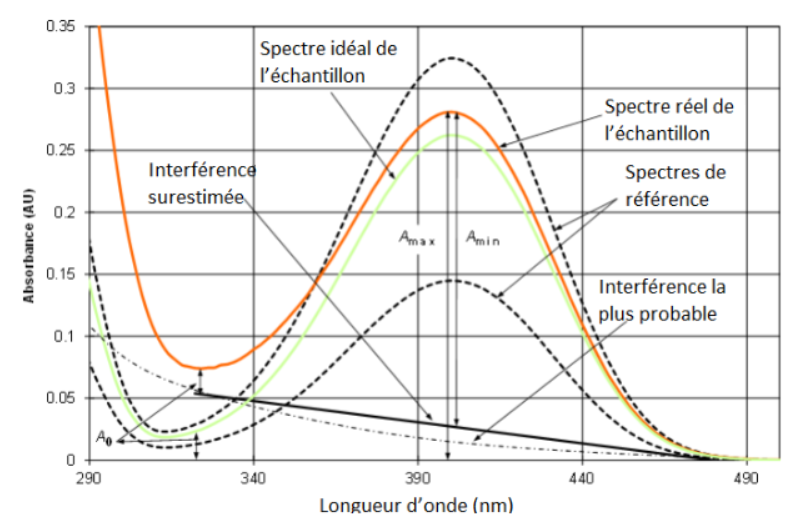
Figure 9.2 : Spectres UV-Visible des solutions de calibration (lignes pointillées), de la solution d'échantillon avec une interférence (rouge), et d'une solution idéale d'échantillon où aucune interférence n'est présente.
Les spectres en pointillés sont ceux de l’analyte dans les solutions de calibration (spectres de référence) où la forme correcte du spectre peut être observée. La ligne verte correspond au spectre “théorique” - c.a.d dépourvu de toute interférence - de la solution d’échantillon. Le spectre le plus probable de l’interférent [3] est présenté par la ligne traitillée. Sa forme suggère que c’est la somme d’un large nombre de différents composés organiques - situation assez commune.
L’absorbance à 400 nm (longueur d’onde de l’absorbance maximum) correspond au signal analytique. Il est évident quand l’on regarde le schéma que si l’on mesure l’absorbance maximale à partir du spectre rouge, on a une absorbance surestimée. Utiliser le spectre vert ou le spectre de l’interférent est impossible, car aucun des deux n’est accessible.
Dans cette situation, on peut essayer d’estimer les valeurs probables d’absorbance maximum et minimum (A) de l’analyte, correspondant respectivement à l’interférence minimale et à la maximale. Une bonne estimation du maximum de A est Amax (cela correspond à la situation très peu probable qu’il n’y a pas d’interférence à 400 nm et que tout le A est dû à l’analyte). Pour estimer le minimum de A on émet l’hypothèse qu’il y a une relation linéaire entre l’effet interférent et la longueur d’onde, comme présenté par la ligne en trait plein nommée “Interférence surestimée”. Elle est surestimée au sens où l’absorbance à 400 nm est plus grande que l’interférence probable. Cette ligne est définie comme suit. Elle est fixée à 0 à 480 nm (il est clair qu’il n’y a pas d’interférence à cette longueur d’onde). A 322 nm la ligne est fixée de manière à ce que la différence d'absorbance du spectre avec l’interférence est la même que la différence du spectre (estimé) sans interférence avec le 0 à la longueur d’onde 322 nm (notée A0 toutes les deux sur le schéma).
A partir des valeurs de Amin et Amax , la valeur d’absorbance corrigée (à utiliser pour le calcul du résultat), ainsi que sa composante de l’incertitude liée à l’interférence peuvent être déterminées comme suit :
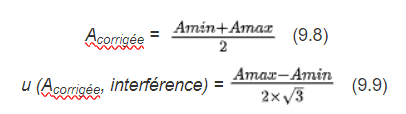
Cette approche est approximative mais son avantage est qu’elle peut être utilisée dans le cas où on ne connaîtrait pas les interférents.
2- Utiliser les données d’interférences de précédentes études.
Si la validation inclut des études d’interférences et que celles-ci sont quantitativement décrites alors ces données peuvent être utilisées pour corriger le résultat et obtenir l’estimation de l’incertitude liée aux possibles interférences. A titre d’exemple, intéressons nous aux valeurs de sélectivité dans la méthode standard ISO 7150:1984 de la détermination spectrophotométrique de la concentration en azote sous forme ammonium. Le tableau 9.2 présente les données concernant l’influence des interférents sélectionnés.
Tableau 9.2 : Influence d’interférents sélectionnés sur la détermination de la concentration en azote sous forme ammonium selon l’ISO 7150:1984 (données tirées de l’ISO 7150:1984).
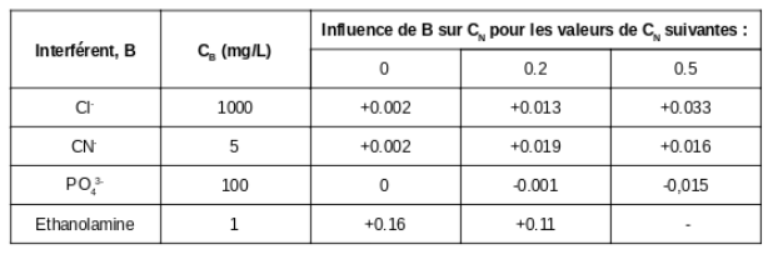
Prenons l’interférence liée au chlore comme exemple. Supposons que nous savons que notre échantillon peut contenir du chlore dans une quantité certainement inférieur à 800 mg/L et que nous avons déterminé une valeur de CN (concentration en azote) d’environ 0.2 dans l’échantillon. Dans ce cas l’interférence maximale serait 0.0013 x 800 / 1000 = 0.0104 mg/L. Si l’on ne connaît pas la quantité de chlore dans l’échantillon et si la déterminer séparément s’avère peu pratique, alors nous pouvons faire l’hypothèse que la concentration en chlore est de (400 ± 400) mg/L. Cette gamme d’incertitude englobe toute la plage de concentrations de 0 à 800 mg/L. L’interférence correspondant à la teneur en chlore serait de +0.0052 mg/L et cette valeur peut être utilisée pour corriger la valeur de CN obtenue (en la soustrayant à CN). Son incertitude type, si on suppose une distribution rectangulaire, est de 0.0054 / 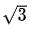 = 0.0030 mg/L. Cette incertitude devrait être incluse additionnellement dans les contributions à l’incertitude.
= 0.0030 mg/L. Cette incertitude devrait être incluse additionnellement dans les contributions à l’incertitude.
3- Utiliser les données sur les interférences via une équation modèle.
Parfois le modèle de la mesure peut directement être utilisé pour prendre en compte les interférences. Un exemple de cette situation peut être retrouvé dans le Test 9.9C.
Autoévaluation sur cette partie du cours : Test 9.5
***
[1] Le même exemple rigoureusement résolu est disponible sur http://www.ut.ee/katsekoda/GUM_examples/. Regardez l’exemple “Ammonium by Photometry” avec le niveau d’élaboration “High (uncertainty estimated at full rigor, suitable for experts)” s’il vous plaît. Comparaison des incertitudes-type composée obtenues : 0,00686 obtenue ici (section 9.7) et 0,0065 mg/L obtenue en toute rigueur. Cela montre que cette approche est acceptable, plus particulièrement car elle mène à une surestimation de l’incertitude plutôt qu’à une sous-estimation de celle-ci.
[2] Si l’interférence est vraiment forte pour une matrice spécifique alors la procédure d’analyse devrait être modifiée et re-validée.
[3] Cette forme du spectre est observée approximativement dans la plupart des cas, si de nombreux composés différents sont présents, chacun à un niveau très faible. De plus, la manière dont le spectre de l’analyte est déformé implique une forme du spectre de l’interférent telle que celle décrite.