MOOC Estimation des incertitudes de mesure en analyse chimique
| Site: | Moodle - Université Claude Bernard Lyon1 |
| Cours: | Estimation des incertitudes de mesure en analyse chimique |
| Livre: | MOOC Estimation des incertitudes de mesure en analyse chimique |
| Imprimé par: | Visiteur anonyme |
| Date: | lundi 13 juillet 2026, 10:39 |
Table des matières
- 1. Introduction
- 2. Le concept d'incertitude de mesure
- 3. L'origine de l'incertitude de mesure
- 4. Les concepts et outils fondamentaux
- 5. Première quantification de l'incertitude
- 6. Principe de l'estimation de l'incertitude de mesure
- 7. Retour sur les effets aléatoires et systématiques
- 8. Fidélité, justesse, exactitude
- 9. L'approche par modélisation ISO GUM
- 10. Aperçu des approches d'estimation de l'incertitude de mesure
- 11. Approche par validation dite de laboratoire unique
- 12. Comparaison des approches
- 13. Comparaison de résultats de mesure
- 14. Autres ressources
- 15. Autotests
1. Introduction
https://sisu.ut.ee/measurement/uncertainty
Ce cours est un cours d'introduction à l'estimation de l'incertitude de mesure, spécifiquement lié à l'analyse chimique (chimie analytique). Le cours présente les concepts principaux et les outils mathématiques d’estimation de l’incertitude de mesure et présente deux approches principales pour l’estimation de cette incertitude de mesure : l’approche de modélisation ISO GUM (approche «bottom-up» ou approche par modélisation) et l'approche par validation par un seul laboratoire telle que mise en œuvre par Nordtest (approche «top-down» ou Nordtest). Le cours contient des vidéos, des exercices pratiques et de nombreux tests d'auto-évaluation.
http://www.uttv.ee/naita?id=17710
| Hello and welcome to the course estimation of measurement uncertainty in chemical analysis. Chemical analysis is almost ubiquitous in nowadays society. Its high importance ranges from high tech production to environmental protection from health care to food safety. We can say without any exaggeration that each and every one of us is affected by chemical analysis. And chemical analysis actually can also be regarded as chemical measurement. So, the essence of chemical analysis is measuring the content of some compounds in some objects. And every measurement always has an uncertainty meaning the measurement result cannot be regarded as absolutely true, absolutely precise. There's always some imprecision some uncertainty about the measurement result. And this measurement uncertainty is one of the most important quality characteristics of any measurement result consequently also of chemical analysis results. And the estimation of measurement uncertainty in chemical analysis is the topic of this course. | Bonjour et bienvenue dans ce cours sur l'estimation de l'incertitude de mesure en analyse chimique. L'analyse chimique est omniprésente dans notre société. Sa grande place va des technologies de production à la protection de l'environnement en passant par la santé et la sécurité alimentaire. On peut dire sans exagération que chacun d'entre nous est impacté par l'analyse chimique. L'analyse chimique peut aussi être vue comme étant la mesure chimique. Ainsi, l'essence même de l'analyse chimique réside dans la mesure de la quantité de composés dans des objets. Et chaque mesure a toujours une incertitude, ce qui signifie que le résultat de la mesure ne peut pas être considéré comme absolument juste et absolument fidèle. Il y a toujours une certaine imprécision, une certaine incertitude sur le résultat de la mesure. Et cette incertitude de mesure est une des caractéristiques de qualité les plus importantes de tout résultat de mesure et par conséquence de tout résultat d'analyse chimique. L'estimation de l'incertitude de mesure en analyse chimique est le sujet de ce cours. |
Malgré son caractère introductif, ce cours a pour objectif d'apporter suffisamment de connaissances et de compétences pour effectuer une estimation de l’incertitude de la plupart des analyses chimiques courantes en laboratoire. Les exemples et les exercices font référence au titrage acide-base, à la détermination de l'azote de Kjeldahl, à la spectrophotométrie UV-Vis, à la spectroscopie d'absorption atomique et à la spectrométrie de masse couplée à la chromatographie en phase liquide (LC-MS). Il est cependant important de souligner que, pour réussir l'estimation de l'incertitude de mesure, l'expérience (en chimie analytique en tant que telle et en estimation de l'incertitude) est cruciale et que celle-ci ne peut être acquise que par la pratique.
Le contenu de ce cours peut également être utile aux personnes qui n’ont pas l’intention de suivre le cours dans son intégralité mais qui souhaitent uniquement trouver des réponses à certaines questions spécifiques. Ce cours a été décrit dans l'article I. Leito, I. Helm, L. Jalukse. Using MOOCs for teaching analytical chemistry: experience at University of Tartu. Anal. Bioanal. Chem. 2015, DOI: 10.1007/s00216-014-8399-y.
Prérequis
Si une connaissance de base de la chimie analytique est seulement nécessaire, une connaissance plus approfondie de la chimie analytique et une connaissance introductive de la statistique mathématique sont un avantage. L'usage d'un logiciel type tableur (MS Excel, OpenOffice, etc.) est fortement recommandé.
http://www.uttv.ee/naita?id=17711
|
Why is measurement uncertainty important? Why do we need to know how to estimate measurement uncertainty? Let us look at an example and let us look at an example of determination of a pesticide thiabendazole in citrus fruits. And suppose we had two laboratories who have made the analysis. One of those laboratories has obtained the result for a particular lot of some citrus fruits has obtained the result of 4.2 milligrams per kilogram. We have here an axis of concentration and we mark, can mark this 4.2 here on this axis. So now suppose the other laboratory has got somewhat higher result and the result of the other laboratory is 4.7 milligrams per kilogram. What can we see about these results are they in agreement or are they in disagreement? Well at first sight we could say that no well no of course they are in disagreement they are strongly different there's no doubt about that. But reality pesticide determination in food at low levels is a difficult analytical task. And it is very difficult to do it in such a way that the results will be very very accurate. Therefore these results actually have high uncertainties. These uncertainties can be several tens of percent of those values themselves. So that if we put realistic uncertainty estimates on these values, we can see a completely different picture. Let us do that. Both of these measurement results are now characterized by uncertainty of roughly plus minus 0.6 milligrams per kilogram. And we see that actually the value according to this laboratories measurement can be anywhere within this range including here. And the true value according to this laboratories result can be anywhere within this range including here. Therefore, we have actually no reason to say that the results are different. In fact, the results agree very well. So, in order to compare two measurement results, we need to know their uncertainties. Now why do people carry out pesticide determination in food? They carry this analytical determination out because they want to know whether we can eat the food or not. So, in order to say whether this thiabendazole concentration in citrus fruit is okay or not, we need to know the maximum permissible limit or maximum residual limit of thiabendazole and this limit in the European Union is five milligrams per kilogram. So here comes this maximum residual limit, and if we now look at these results, we can see that although the results themselves are below 5 milligrams per kilogram there is also some probability that the true value is above there. Probably the true content of pesticide in the citrus fruit is below 5 milligrams per kilogram but we cannot be completely sure in this and the decision remains with the respective Food Authority as to whether these fruits could still be sold or not. So, we see that measurement uncertainty is first of all an important quality characteristic content measurement result and secondly it also is necessary for comparing results. And this has been recognized by different authorities and in fact nowadays one of the requirements if a laboratory wants to accredit its management system is the ability to be able to estimate measurement uncertainty for the results of chemical analysis and measurements that laboratory makes. So that the measurement uncertainty estimation ability is also required from the laboratories by authorities. |
Pourquoi l'incertitude de mesure est-elle importante ? Pourquoi avons-nous besoin d'estimer l'incertitude d'une mesure ? Regardons un exemple et prenons l'exemple de la détermination d'un pesticide, la thiabendazole dans des citrons. Supposons que nous ayons deux laboratoires qui ont réalisé cette analyse. Un des laboratoires a obtenu un résultat pour un lot de citrons, et a obtenu un résultat de 4,2 milligrammes par kilogramme. Nous avons ici un axe de concentration et nous pouvons placer 4,2 sur cet axe. Maintenant supposons que l'autre laboratoire a obtenu un résultat plus grand et le résultat de cet autre laboratoire est de 4,7 milligramme par kilogramme. Ce que l'on peut voir au sujet de ces résultats : sont-ils en accord ? Ou sont-ils en désaccord ? Au premier coup d'œil nous pouvons dire que bien sûr ils sont en désaccord, ils sont très différents, il n'y a aucun doute là-dessus. Mais en réalité, la détermination des pesticides dans la nourriture à très faible niveau est une analyse très difficile. Et il sera très difficile de le faire de telle sorte que les résultats soient extrêmement exacts. Ainsi, ces résultats sont actuellement assortis d'une grande incertitude. Cette incertitude peut représenter plusieurs dizaines de pourcents de ces valeurs. Et si nous plaçons des valeurs réalistes de l'incertitude sur ces valeurs, nous pouvons avoir une image complètement différente. Faisons le… Les deux résultats de mesure sont maintenant caractérisés par une incertitude de plus ou moins 0,6 milligrammes par kilogramme. Nous voyons maintenant que la valeur attribuée à cette mesure par le laboratoire peut se situer n'importe où dans cette plage, même ici. Et la valeur vraie selon les résultats de ce laboratoire peut être n'importe où dans cette plage, même ici. Ainsi, nous n'avons aucune raison pour dire que ces résultats sont différents. En fait, les résultats sont en très bon accord. Donc pour comparer deux résultats de mesure, nous avons besoin de connaitre leurs incertitudes. Pourquoi les gens recherchent-ils des pesticides dans l'alimentation ? Ils réalisent cette mesure analytique parce qu'ils veulent savoir si nous pouvons manger ces aliments ou pas. Pour dire si cette concentration en thiabendazole dans les citrons est satisfaisante ou pas, nous avons besoin de connaitre la limite maximale autorisée ou limite maximale résiduelle de thiabendazole, et cette limite dans l'Union Européenne est de 5 milligrammes par kilogramme. Apparaît maintenant cette limite maximale résiduelle, et si nous regardons maintenant ces résultats nous pouvons voir que si les résultats sont inférieurs à 5 milligrammes par kilogramme, il existe une certaine probabilité que la valeur vraie soit au-dessus. Probablement, la valeur vraie en pesticide dans les citrons est inférieure à 5 milligrammes par kilogramme mais nous ne pouvons en être totalement certains. La décision incombe à l'autorité de surveillance de l'alimentation de décider si ces fruits peuvent être vendus ou pas. Ainsi, l'incertitude d'une mesure est tout d'abord une caractéristique importante de la qualité du résultat de mesure et d'autre part, elle est nécessaire pour comparer des résultats. Ceci a été reconnu par différentes autorités et, en fait maintenant, une des exigences pour un laboratoire qui souhaite accréditer son système de management réside dans sa capacité à estimer les incertitudes de mesure pour les résultats d'analyse chimique et les mesures qui sont réalisées par le laboratoire. Ainsi l'aptitude à estimer l'incertitude de mesure est demandée aux laboratoires par les autorités. |
2. Le concept d'incertitude de mesure
https://sisu.ut.ee/measurement/introduction-concept-measurement-uncertainty
Résumé : Cette section présente les concepts de mesurande, valeur vraie, valeur mesurée, erreur, incertitude de mesure et probabilité.
http://www.uttv.ee/naita?id=17583
|
Let us see now what measurement uncertainty is and then let us see this on an example of a glass of water. Water is the most important liquid in our lives we all drink a lot of water and it is very important that drinking water is healthy, and one part of its healthiness comes from the content of minerals. There are always some small amounts of salts in this water. Many of those salts are even somewhat good for our health but some however are toxic. And if we speak about toxic minerals or toxic salts, we first of all speak about different metal cations. And let us examine the toxic metal lead. There's always some small amount of lead in drinking water but as long as it's small enough it's not really harmful for our health. But it's important that the small level it does not get too high and therefore it’s important to make chemical analysis of water to determine whether it is suitable for drinking or not. So, our measurement in this case is lead content in water and we denote it by C, and we measure it in micrograms per liter. This concentration can assume different values therefore we have here an axis concentration axes and in the in order to make this example more concrete let us also put some real concentration values on this axis. So, this is five, this is six, this is seven micrograms per liter. Now if we again take this glass of water then obviously there's a certain number of lead ions swimming around in this water. Meaning there is some true concentration of lead in this water, and if we were able to count those layered ions, we would know how much they are. Unfortunately, and this is true for almost all analytical chemistry we almost never can really count molecules or atoms meaning the true value will almost always remain unknown for us. And from the point of view of measurement science we can say that the true value is an abstract concept. So even though we are always aiming at the true value with our measurement, we almost never can really achieve it. Let us denote this true value here now. Let's assume it's somewhere here so we call it Ctrue. And according to this scale it's equal to 5.7 micrograms per liter. Now we optimize our measurement procedures in such a way that we would get our measurement result as close to the true value as possible, but we almost never exactly achieve it and we almost never will know if we achieved it or not. So now let us suppose that our measured value in this case happens to be six point one micrograms per litter. Now the difference between the true value and the measured value is called error. And we denote error by Delta, and delta is equal to Cmeasured minus Ctrue. Now, as true value for us is an abstract concept so is also the error because if we would know what the error is then by obviously knowing our measured value, we would be able to calculate the true value, but this is not possible. So unfortunately, people cannot use the concept of error or true value for characterizing the quality of this measured value here. So, what people do instead, they define a range around this measured value and they define this range in such a way that the true value would be within this range with high probability. So, and this range is called uncertainty range. And the half width of this range is called uncertainty. We can be noted by U and in this case the uncertainty is equal to 0.5 micrograms per litter. Now if we have made this measurement and obtained this measurement result and we have made uncertainty estimation taking into account all the uncertainty sources which are influencing the measurement result and we arrived at this uncertainty estimate, we can present our measurement result. And this would look like this. Result, our C is equal 6.1 plus minus 0.5 micrograms per liter. So, instead of trying to give the exact true value or the difference from the true value which we cannot do, we give this range, and this range means that with high probability the true value lies within this range. So, this result tells us to a person that looks at this result that the true value of lead in this water is between 5.6 and 6.6 micrograms per liter. And as we see in this particular case it fully holds because the true value is 5.7 It would be fair to ask why am I speaking about high probability, why don't I say that the true value certainly lies within region? We will see later on that almost never we can present uncertainty in such a way that it embraces the true value with 100% probability. Almost always uncertainty presentation is probabilistic. We will see later on that this way of writing is actually not enough if we want to correctly present measurement result, but that will come later and for the moment this way of presenting the result is for this example sufficient. It is interesting to ask now as we have received this kind of result, can we drink this water or not? Is this level of lead in drinking water in the permissible region or not? The permissible value of lead concentration in drinking water in the European Union is 10 micrograms per liter. We see that 6.1 of course is significantly below 10 but we also must take into account the uncertainty, meaning in principle the true value would also be somewhere higher here. As I said uncertainty range never can have 100 percent probability but it is good to know that probability outside of this range decreases exponentially so that with very high probability the value is within this range, and if we already would go to 5 to 7 range the probability would be extremely high so that the probability of the true content being somewhere above there is very very low. So that this this water is fully suitable for drinking! |
Regardons maintenant ce que représente l'incertitude de mesure. Regardons ceci sur l'exemple d'un verre d'eau. L'eau est le liquide le plus important pour notre vie, nous buvons énormément d'eau et il est très important que cette eau soit saine. Une partie de sa qualité sanitaire provient du fait qu'elle contient des minéraux. Il y a toujours de petites quantités de sel dans l'eau. La plupart de ces sels sont même nécessaires pour notre santé, mais quelquefois ils sont toxiques. Et si nous parlons de minéraux ou de sels toxiques, nous devons parler des différents cations métalliques. Examinons un métal toxique tel que le plomb. Il y a toujours une petite quantité de plomb dans l'eau de boisson, mais pour autant qu'elle soit suffisamment faible, elle n'est pas toxique pour notre santé. Mais il ne faut pas que ce niveau devienne trop important, et donc il est important de réaliser une analyse chimique de l'eau pour déterminer si elle est acceptable comme boisson ou pas. Ainsi, dans ce cas, notre mesure est la quantité en plomb dans l'eau que nous nommerons C et nous le mesurerons en microgramme par litre. Cette concentration peut prendre différentes valeurs donc nous avons ici un axe : l'axe des concentrations, et dans le but de rendre cet exemple plus concret, nous allons positionner la valeur réelle de la concentration sur cet axe. Nous avons ainsi cinq, six et sept microgrammes par litre. Maintenant, si nous prenons ce verre d'eau il y a évidemment un certain nombre d'ions plomb en suspension dans cette eau. Cela signifie aussi qu'il y a une vraie concentration de plomb dans l'eau et si nous sommes en capacité de compter ces ions, nous pourrions connaitre leur nombre. Malheureusement, et ceci est vrai pour presque toutes les analyses chimiques, nous ne pouvons pas réellement compter les molécules ou atomes, ce qui signifie que la vraie valeur restera quasiment toujours inconnue pour nous. Et du point de vue de la science de la mesure, nous pouvons dire que la valeur vraie est un concept abstrait. Ainsi, même si nous tendons vers la valeur vraie avec nos mesures, nous ne pouvons presque jamais l'atteindre. Plaçons cette valeur vraie. Nous supposerons qu'elle est ici et nous l'appellerons Cvraie (Ctrue). Et sur cette échelle, elle est égale à 5,7 microgrammes par litre. Maintenant, nous optimisons nos procédures de mesure de telle sorte que nous ayons notre résultat de mesure aussi proche que possible de la valeur vraie, mais nous ne pourrons jamais l'atteindre exactement, et nous ne saurons jamais si nous l'atteignons ou pas. Maintenant, supposons que notre valeur mesurée soit égale à 6,1 microgrammes par litre. La différence entre la valeur vraie et la valeur mesurée est appelée erreur. Et nous désignerons cette erreur par Delta et Delta est égal à Cmesurée moins Cvraie. Maintenant, comme la valeur vraie est un concept abstrait, l'erreur l'est aussi parce que si nous connaissions la valeur de l'erreur, alors en connaissant la valeur mesurée, nous serions à même de calculer la valeur vraie. Mais ce n'est pas possible. Malheureusement, on ne pourra pas utiliser le concept d'erreur ou de valeur vraie pour caractériser la qualité de la valeur mesurée ici. Ainsi, ce que les gens font à la place, ils définissent une étendue autour de la valeur mesurée, et ils définissent une étendue de telle sorte que la valeur vraie se trouve dans cette étendue avec une grande probabilité. Ainsi, cette étendue est appelée étendue d'incertitude. La demi-largeur de cette étendue est appelée incertitude. Nous pouvons la noter U et, dans ce cas, l'incertitude est égale à 0,5 microgramme par litre. Si nous avons réalisé cette mesure et obtenu un résultat de mesure, et si nous avons réalisé une estimation de l'incertitude en tenant compte de toutes les sources d'incertitude qui influencent le résultat de la mesure et que nous arrivions à une valeur de l'incertitude, nous pourrons présenter notre résultat de mesure. Et il ressemblera à ceci. Résultat : C est égale à 6,1 0.5 microgramme par litre. Ainsi, plutôt que d'essayer de donner la valeur exacte de la valeur vraie ou la différence entre la valeur vraie ce que nous ne pouvons faire, nous donnons une étendue et cette étendue signifie, qu'avec une grande probabilité, la valeur vraie est dans cette étendue. Ainsi, ce résultat signifie que pour une personne qui regarde ce résultat, que la valeur vraie du plomb dans l'eau est comprise entre 5,6 et 6,6 microgrammes par litre. Et comme nous pouvons le voir dans ce cas particulier ceci est correct car la valeur vraie est 5,7. Il pourrait être normal de demander pourquoi je parle de grande probabilité et pourquoi je ne dis pas que la valeur vraie est certainement comprise dans cette région. Nous verrons plus tard que nous ne pourrons quasiment jamais donner une incertitude de telle sorte qu'elle contienne la valeur vraie avec une probabilité de 100%, Presque toujours, l'expression de l'incertitude est de type probabiliste. Nous verrons plus tard que cette façon d'écrire n'est pas suffisante si nous voulons présenter correctement le résultat de la mesure, mais ceci sera présenté plus tard et pour le moment cette façon de présenter le résultat et suffisant pour cet exemple. Il est intéressant de se demander maintenant que nous avons reçu ce type de résultat, peut-on boire cette eau ou pas ? Est-ce que ce niveau de plomb dans l'eau est admis dans la région ou pas ? La valeur autorisée de la concentration en plomb dans l'eau de boisson de l'Union Européenne est de 10 mg/L. Nous voyons que 6,1 est bien significativement inférieur à 10 mais nous devons tenir compte de l'incertitude, ce qui signifie en principe que la valeur vraie pourrait être plus grande ici. Comme je l'ai dit, l'étendue de l'incertitude ne peut pas avoir une probabilité de 100% mais il faut savoir que la probabilité hors de cette étendue décroit exponentiellement. Et ainsi, avec une très grande probabilité la valeur vraie est à l'intérieur de cette étendue qui va de 5 à 7. Cette probabilité étant très importante, la probabilité que la vraie valeur de la concentration soit quelque part plus grande est très très faible. Ainsi, cette eau est totalement buvable ! |
La mesure est un processus permettant d'obtenir expérimentalement la valeur d'une quantité. La quantité que nous avons l'intention de mesurer s'appelle le mesurande. En chimie, le mesurande est généralement la quantité de matière ou la concentration d'une entité chimique (molécule, élément, ion, etc.) dans un objet. L'entité chimique à déterminer est appelée analyte. Les mesures chimiques peuvent être, par exemple, la concentration de plomb dans un échantillon d’eau, la teneur en pesticide du thiabendazole dans une orange ou la teneur en matière grasse dans une bouteille de lait. Dans l'exemple précédent, le plomb (élément), l'acide ascorbique (molécule) et le tissu adipeux (groupe de molécules différentes) sont les analytes. L'eau, l'orange et le lait sont des objets d'analyse (ou des échantillons prélevés à partir d'objets d'analyse).
En principe, le but d’une mesure est d’obtenir la vraie valeur du mesurande. Tous les efforts sont faits pour optimiser la procédure de mesure (procédure d'analyse chimique ou procédure d'analyse) de manière à ce que la valeur mesurée soit aussi proche que possible de la valeur réelle. Cependant, notre résultat de mesure ne sera qu'une estimation de la valeur réelle et la valeur réelle restera (presque) toujours inconnue de nous. Par conséquent, nous ne pouvons pas savoir exactement à quel point notre valeur mesurée est proche de la valeur réelle - notre estimation comporte toujours une certaine incertitude.
La différence entre la valeur mesurée et la valeur vraie est appelée erreur. Une erreur peut avoir un signe positif ou négatif. L’erreur peut être considérée comme composée de deux parties - l’erreur aléatoire et l’erreur systématique - qui seront traitées plus en détail dans les prochains chapitres. Comme la valeur vraie, l'erreur nous est inconnue. Par conséquent, elle ne peut pas être utilisée dans la pratique pour caractériser la qualité de notre résultat de mesure, son accord avec la valeur réelle.
La qualité du résultat de mesure est caractérisée par l’incertitude de mesure (ou simplement l’incertitude), qui définit un intervalle autour de la valeur mesurée CMEASURED, où la valeur vraie CTRUE se situe avec une certaine probabilité. L'incertitude de mesure U elle-même est la demi-largeur de cet intervalle et est toujours non négative. Le schéma suivant (similaire à celui de la vidéo) illustre ceci :
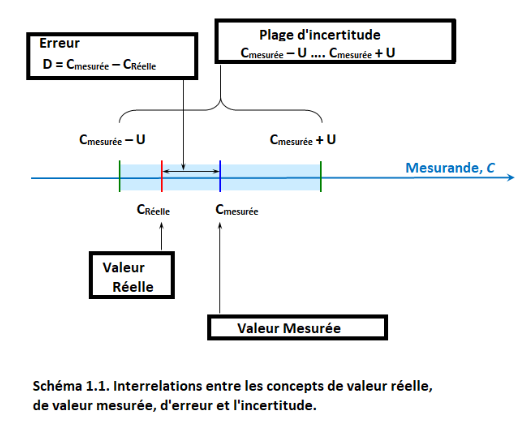
L’incertitude de mesure est toujours associée à une probabilité - comme on le verra dans les prochains chapitres, il n’est généralement pas possible de définir l’intervalle d’incertitude de telle sorte que la valeur réelle y soit comprise avec une probabilité de 100%.
L'incertitude de mesure, telle qu'exprimée ici, s'appelle dans un contexte également l'incertitude de mesure absolue. Cela signifie que l'incertitude de mesure est exprimée dans les mêmes unités que le mesurande. Comme nous le verrons dans les cours suivants, il est parfois plus utile d’exprimer l’incertitude de mesure sous la forme d’une incertitude relative de mesure, qui est le rapport entre l’incertitude absolue Uabs et la valeur mesurée y :
 (1.1)
(1.1)
L'incertitude relative est une quantité sans unité, qui est parfois exprimée en pourcentage.
L’incertitude de mesure est différente de l’erreur en ce sens qu’elle n’exprime pas de différence entre deux valeurs et qu’elle n’a pas de signe. Par conséquent, ell ne peut pas être utilisée pour corriger le résultat de la mesure et ne peut pas être considérée comme une estimation de l'erreur car celle-ci a un signe. Au lieu de cela, l’incertitude de mesure peut être considérée comme notre estimation de la différence absolue la plus probable entre la valeur mesurée et la valeur réelle. Avec une probabilité élevée, la différence entre la valeur mesurée et la valeur réelle est en fait inférieure à l'incertitude de mesure. Cependant, il existe une faible probabilité que cette différence puisse être supérieure à l'incertitude de mesure.
Autoévaluation sur cette partie du cours : Test 1
3. L'origine de l'incertitude de mesure
https://sisu.ut.ee/measurement/origin-measurement-uncertainty
Résumé : Présentation, sur l'exemple du pipetage, de l'origine de l'incertitude de mesure. Le concept de sources d'incertitude (effets provoquant un écart de la valeur mesurée par rapport à la valeur réelle) est présenté. Les principales sources d’incertitude liées au pipetage sont introduites et expliquées : répétabilité, étalonnage, effet de la température. Une explication des effets aléatoires et systématiques est donnée. Le concept de répétabilité est introduit.
La première vidéo montre comment le pipetage avec une pipette volumétrique classique est réalisé et explique d'où provient l'incertitude du volume pipeté.
Pourquoi les résultats de mesure présentent-ils une incertitude ? Le concept de source d'incertitude expliqué sur l'exemple du pipetage
http://www.uttv.ee/naita?id=17577
| Let us see now where measurement uncertainty comes from, and let us see that on the example of pipetting. So I am holding in my hand a 10 milliliter volumetric pipet and I'm going to pipet now with this pipette exactly 10 milliliters of water from this water beaker into this sample beaker. And suppose we need this water for some kind of chemical analysis and we need that in the sample beaker we really have 10 milliliters. And I'm going to conduct this pipetting now and then I'm going to explain what are the possible uncertainty sources that come into play when we do pipetting. So the pipette is a clean pipette, but even in the case of a clean pipette it is a good idea to first rince it a couple of times with the solution that we are going to pipette. So, we fill the pipette. It is not important at the moment to fill it exactly to the mark Instead we deliberately overfill and we empty it into the waste beaker. It is important to say that rinsing with the solution that you are going to pipette, which is water in our case, is much more useful than rinsing with pure water. Actually rinsing with pure water cannot be recommended because if the solution itself is not pure water, then that rinsing will possibly dilute your solution which eventually you will be pipeting. So I'm now rinsing two times not taking special care to fill it to any special level and now I'm going to fill it to exactly 10 milliliters. Again I'm filling the pipette and I'm going to first overfill a little bit. And then the remaining water, I will drain into the same waste beaker and now it is important that I keep the mark exactly at the height of my eyes and that I keep the pipette really vertical. All right. And now, I drain the remaining water into the sample beaker and again it is important to keep the pipette vertically. And also, as you can see, I'm holding the pipette tip against the wall of the beaker. And, after all the liquid has been pipeted, I wait for five seconds before removing the pipette. So keeping the pipette tip against the beaker wall is very important because there's always some small amount the water that remains in the volumetric pipet and it should remain there. But some water still comes from the walls and it will be each time aspirated into the beaker the same way if you keep the pipette tip against the beaker wall not in the air. Alternatively you can also keep the pipette tip against the solution surface. So now as we've done this pipeting, the sample beaker now contains as close as we can volume to ten milliliters of water, and let us think now where uncertainty comes from. There are several uncertainty sources that are inherent in pipetting, the first of them is that even though I took every care to fulfill the pipette really to the mark exactly always I either slightly overfill or slightly underfill the pipette. Secondly the mark itself even though it is supposed to be exactly 10 milliliters but this mark position also has an uncertainty which actually is also stated on the pipette. It's plus minus 0.03 milliliters in this particular case. Then the third uncertainty component is the uncertainty due to the temperature effect. Temperature affects both pipette volume and water density. The volume of glassware is not very sensitive towards temperaturen but the volume of liquid is. And therefore if the temperature is above or below the temperature at which the pipette was calibrated, we get respectively less or more water molecules in our beaker. And almost all glassware always is calibrated at 20 degrees centigrade which also is the case of this particular pipette as it can be seen it reads 20 degrees centigrade. These three uncertainties always need to be taken into account when we look at uncertainty of volumetric measurement, but there can be other uncertainty sources which sometimes can be of importance. Almost all these volumetric glassware are calibrated using water, therefore they are meant to deliver liquid that really is very much like water meaning either water or dilute solution. Whenever you pipette oil or some very viscous solution, maybe some high density solution, then the pipetted volume can differ and then it's a good idea to pipette, to calibrate the pipette himself or herself. And finally if the pipette is not clean then there can remain droplets of water on pipette walls after pipetting which means that the volume delivered is not anymore the same as we need. As we see in this our case there are no water droplets so this uncertainty source in our case does not come into play. | Regardons maintenant d'où vient l'incertitude de mesure, et regardons cela au travers de l'exemple du pipetage. Je tiens dans ma main une pipette de 10 millilitres et je vais maintenant pipeter avec cette pipette exactement 10 millilitres d'eau, depuis ce bécher d'eau vers le bécher échantillon (sample). Supposons que nous ayons besoin de cette eau pour une analyse chimique, et que nous ayons besoin d'avoir dans le bécher échantillon réellement 10 millilitres d'eau. Je vais maintenant réaliser le pipetage et j'expliquerai quelles sont les sources possibles d'incertitude qui sont impliquées lorsque nous pipetons. Cette pipette est une pipette propre, mais même avec une pipette propre, c'est une bonne idée de tout d'abord la rincer une paire de fois avec la solution que nous allons ensuite pipeter. On remplit donc la pipette. Il n'est pas important pour le moment de la remplir jusqu'au trait. En fait, nous remplissons au-delà du trait et nous la vidons dans le bécher poubelle. Il est important de préciser que rincer avec la solution que l'on doit prélever, qui est de l'eau dans notre cas, est beaucoup plus utile que rincer avec de l'eau pure. Dans ce cas, rincer avec de l'eau pure n'est pas recommandé parce que la solution à analyser n'est pas de l'eau pure. Et ce rinçage à l'eau pure pourrait diluer la solution que vous voulez pipeter. Je rince maintenant deux fois sans précaution spécifique pour remplir à un niveau particulier, et je vais maintenant remplir pour avoir exactement 10 millitres. Je remplis la pipette et je vais tout d'abord la remplir au-delà du trait. Concernant l'excès d'eau, je vais le verser dans le même bécher poubelle. Maintenant il est important que je mette le trait exactement au niveau de mes yeux et que je garde la pipette parfaitement verticale. Voilà. Je verse l'eau dans le bécher échantillon et à nouveau il est important de garder la pipette verticale. Et aussi, comme vous pouvez voir, je positionne la pointe de la pipette contre la paroi du bécher (NDT angle de 45°). Et, après que le liquide ait été pipeté, j'attends 5 secondes avant de retirer la pipette. Garder la pointe de la pipette contre la paroi du bécher est très important car il y a toujours une petite quantité d'eau qui reste dans la pipette, et qui doit rester. Mais un peu d'eau vient encore des parois et elle sera aspirée chaque fois dans le bécher de la même manière si vous gardez l'embout de la pipette contre la paroi du bécher et non dans l'air. Vous pouvez aussi garder l'embout de la pipette contre la surface de la solution. Maintenant que nous avons fait ce pipetage, le bécher d'échantillon contient aussi près que possible un volume de dix millilitres d'eau. Réfléchissons maintenant d'où vient l'incertitude. Il y a plusieurs sources d'incertitude inhérentes au pipetage. La première d'entre elles est que même si j'ai pris le plus grand soin pour remplir la pipette exactement au trait de jauge, je remplis toujours légèrement trop ou légèrement moins la pipette. Deuxièmement, le trait de jauge lui-même, même si il est censé être exactement à 10 millilitres, cette position du trait a aussi une incertitude qui est également indiquée sur la pipette. C'est plus moins 0,03 millilitre dans ce cas particulier. Enfin, la troisième composante d'incertitude est l'incertitude due à l'effet de la température. La température affecte à la fois le volume de la pipette et la densité de l'eau. Le volume de la verrerie n'est pas très sensible à la température mais le volume du liquide l'est. Et donc, si la température est supérieure ou inférieure à la température à laquelle la pipette a été calibrée, nous obtenons respectivement moins ou plus de molécules d'eau dans notre bécher. Presque toute la verrerie est calibrée à 20 degrés Celsius, ce qui est également le cas de cette pipette en particulier, comme on peut le voir, il est indiqué 20 degrés Celsius. Ces trois incertitudes doivent toujours être prises en compte lorsque nous examinons l'incertitude de la mesure volumétrique, mais il peut y avoir d'autres sources d'incertitude qui peuvent parfois être importantes. Presque toutes ces verreries volumétriques sont calibrées à l'eau, donc elles sont destinées à délivrer un liquide qui ressemble beaucoup à de l'eau, c'est-à-dire de l'eau ou une solution diluée. Chaque fois que vous pipetez de l'huile ou une solution très visqueuse, peut-être une solution à haute densité, alors le volume pipeté peut différer et c'est une bonne idée pour pipeter, de calibrer la pipette soi-même. Enfin, si la pipette n'est pas propre, il peut rester des gouttelettes d'eau sur les parois de la pipette après pipetage, ce qui signifie que le volume délivré n'est plus le même, comme nous en avons besoin. Comme nous le voyons dans ce cas-ci, il n'y a pas de gouttelettes d'eau, cette source d'incertitude dans notre cas n'entre pas en ligne de compte. |
La deuxième vidéo montre le pipetage avec une pipette automatique moderne et explique les sources d'incertitude associées au pipetage avec une pipette automatique.
Source d'incertitude de mesure du pipetage avec une pipette automatique
http://www.uttv.ee/naita?id=18164
| Let us see now where measurement uncertainty comes from if we do pipetting with an automatic pipette. I'm holding in my hand an automatic pipette and with despite that I'm going to pipette now exactly 2,3 milliliters of water from this water beaker into this sample beaker. Let us first examine this pipette a little bit it is a pipette with adjustable volume and the volume can be adjusted from 500 to 5000 microliters meaning from 0.5 to 5 milliliters. And with this knob, I can set the volume here and what I have done now I have exactly said 2,3 milliliters yet what I need to pipette. Before pipetting I need to attach to the pipette, a pipette tip. So and now the pipette is ready for use. Pipetting with an automatic pipette is in several ways different from pipetting with the glass pipette, but some things are similar also. First of all, also with the automatic pipette is useful to rinse before pipetting. But here the rinsing is not so much about cleaning the pipette tip because the tip as they come from such containers or from packages are usually very carefully clean. But rinsing the tip as I will show you in a minute serves the purpose of saturating the gas phase above the liquid with the liquid vapors which is important for pipetting accurate volume. Pipetting with an automatic pipette is done using this knob and it has two stops. One stop and the second stop. If I push it to the first stop then exactly the right volume of the liquid is delivered but the second stop is meant for emptying the pipette completely. So that before taking the liquid into the pipette, I pushed to the first stop and release and when I deliver the liquid I push to the second stop and release. And let's see now how this works. I first rinse the pipette two times and then I will pipette the liquid volume of 2,3 milliliters into this sample beaker. So the pipette is held vertically, and the liquid is aspirated into the pipette slowly. Okay. And now I release it into the waste beaker. And now I push to the second stop. Again I push to the first stop immerse, take the liquid slowly. Okay, And now slowly push to the second stop and I touch with pipette tip either the beaker wall or the liquid itself. And now as I rinse two times I now take the third time the liquid, and I deliver it into the sample beaker. And again I dispense completely and touch with the top of the pipette the liquid. So this beaker now should contain exactly 2,3 milliliters of water and it's important to see now that the pipette tip has to be completely clean of any liquid droplets. This means that all the liquid has been dispensed into that beaker. Let us see now where the uncertainty comes from if we do pipetting with such an automatic pipette. Pipetting with the automatic pipette on one hand looks easier than pipetting with the glass pipette. But in fact it's more tricky and there are more uncertainty sources involved. First of all, again, we have the calibration uncertainty as we had with the glass pipette and this is the calibration accuracy component of this pipette. And it can be said that with with large volume automatic pipettes it usually is significantly higher than with glass pipette but with smaller ones it can be smaller. And secondly it's very important that if a glass pipette is calibrated then it keeps its calibration for very long time almost infinitely. But these pipettes needs occasional recalibration because the mechanics little bit changes with time inside here. And so the calibration can drift away and this drift can be maybe up to 1% of the volume which is quite quite a lot. Secondly an important uncertainty component is the speed of pushing and releasing this button. It has to be as uniform as possible and it's very good if the person who uses it also calibrates it with the same kind of speed. Then the third important uncertainty sources is rinsing and vapor saturation with aqueous solutions. This is not so critical, and in fact in simpler applications people often do pipetting without any rinsing at all. But if you pipette something which is more volatile then such rinsing is absolutely must, otherwise you can get maybe 10 or 15% lower volume than otherwise you should. As with glass pipettes, it is important with automatic pipettes that a liquid which is pipetted is similar to the one with which the pipette was calibrated. As long as we pipette dilute aqueous solutions all is fine. But if the pipette is calibrated with water as is usually the case, and if the solution that then is pipetted is some concentrated salt solution or may be concentrated alkali or acid then again quite significant additional uncertainty can come. And it can come largely because of the density difference of the liquids and the density difference will cause less liquid to be held in this pipette because some slight vacuum will form here so that part of the liquid will fall out before you can take the pipette out of the liquid. Then the temperature effect of course has the same influence here as in the glass pipette but again here it's more tricky. If the pipette is kept in hand for a long time then the temperature of this mechanical part increases and it can be significantly warmer than the temperature in the room. So that in fact the temperature effect with this type of pipette can be larger. There are also some other uncertainty sources that rather refer to sloppy working practices. One of them is using an incorrect tip. The next one is using a tip but not attaching it properly. And then, also if pipetting isn't done either under an angle, this also introduces additional uncertainty. True, in certain applications, for people it is much more easy to pipette at an angle than vertically, and in such a case people oftentimes calibrate the pipette also under angle then it's okay. |
Voyons maintenant d'où vient l'incertitude de mesure si nous pipetons avec une pipette automatique. Je tiens dans ma main une pipette automatique et, avec cela, je vais pipeter exactement 2,3 millilitres d'eau de ce bécher d'eau dans ce bécher à échantillon. Examinons d'abord un peu cette pipette, c'est une pipette à volume réglable et le volume peut être ajusté de 500 à 5000 microlitres soit de 0,5 à 5 millilitres. Et avec ce bouton, je peux régler le volume ici, et c'est ce que je fait maintenant, j'ai exactement dit 2,3 millilitres, c'est ce que j'ai besoin de pipeter. Avant de pipeter, je dois fixer à la pipette un embout de pipette. La pipette est maintenant prête à l'emploi. Le pipetage à l'aide d'une pipette automatique diffère de plusieurs façons du pipetage à la pipette en verre, mais certaines choses se ressemblent aussi. Tout d'abord, avec une pipette automatique, il est également utile de rincer avant le pipetage. Mais ici le rinçage ne consiste pas tant à nettoyer l'embout de la pipette, car l'embout provenant de ces boites ou de leurs emballages a été généralement très soigneusement nettoyé. Mais le rinçage de l'embout, comme je vais vous le montrer dans une minute, sert à saturer la phase gazeuse au-dessus du liquide avec les vapeurs liquides, ce qui est important pour pipeter un volume exact. Le pipetage avec une pipette automatique se fait à l'aide de ce bouton qui a deux butées. Un premier arrêt et un deuxième arrêt. Si je pousse le bouton jusqu'à la première butée, le volume exact du liquide est délivré, et la deuxième butée est destinée à vider complètement la pipette. Ainsi, avant d'introduire le liquide dans la pipette, j'ai poussé jusqu'à la première butée et je l'ai relâché, et lorsque je délivre le liquide, j'ai poussé jusqu'à la deuxième butée et relâché. Et voyons maintenant comment ça marche. Je rince d'abord la pipette deux fois, puis je pipette un volume de liquide de 2,3 millilitres dans ce bécher à échantillon. Ainsi, la pipette est maintenue verticalement et le liquide est aspiré lentement dans la pipette. D'accord. Et maintenant, je le jette dans le bécher à déchets. Et maintenant, je pousse jusqu'au deuxième arrêt. Encore une fois, je pousse jusqu'au premier arrêt, immerge, et prend le liquide lentement. Ok, et maintenant, je pousse doucement jusqu'à la deuxième butée et je touche avec la pointe de la pipette soit la paroi du bécher, soit le liquide lui-même. Et maintenant que j'ai rincé deux fois, je prends la troisième fois le liquide et je le verse dans le bécher d'échantillon. Et encore une fois, je verse complètement et je touche le liquide avec le dessus de la pipette. Ce bécher doit donc maintenant contenir exactement 2,3 millilitres d'eau et il est important de voir maintenant que l'embout de la pipette doit être complètement propre de toute gouttelette de liquide. Cela signifie que tout le liquide a été versé dans ce bécher. Voyons maintenant d'où vient l'incertitude si nous faisons du pipetage avec une telle pipette automatique. Le pipetage avec la pipette automatique d'une part semble plus facile que le pipetage avec la pipette en verre. Mais en fait, c'est plus délicat et il y a plus de sources d'incertitude. Tout d'abord, encore une fois, nous avons l'incertitude d'étalonnage que nous avions avec la pipette en verre et c'est la composante d'exactitude d'étalonnage de cette pipette. Et on peut dire qu'avec les pipettes automatiques de grand volume, elle est généralement beaucoup plus élevée qu'avec les pipettes en verre, mais avec les plus petites, elle peut être plus petite. Et deuxièmement, une fois la pipette en verre calibrée, elle conserve son calibrage pendant très longtemps, presque à l'infini. Or ces pipettes automatiques ont besoin d'un recalibrage occasionnel parce que la mécanique change un peu avec le temps à l'intérieur. Ainsi, l'étalonnage peut dériver et cette dérive peut représenter jusqu'à 1 % du volume, ce qui est assez important. Deuxièmement, un élément important d'incertitude est la vitesse de pression et de relâchement de ce bouton. Elle doit être aussi uniforme que possible. Et c'est très bien si la personne qui l'utilise, l'étalonne aussi avec le même type de vitesse. Ensuite, la troisième incertitude importante provient du rinçage et de la saturation en vapeur des solutions aqueuses. Ceci n'est pas si critique, et en fait, dans les applications les plus simples, les gens font souvent du pipetage sans aucun rinçage. Mais si vous pipetez quelque chose qui est plus volatil, alors un tel rinçage est absolument nécessaire, sinon vous pouvez obtenir peut-être 10 ou 15% de volume en moins que ce que vous devriez obtenir autrement. Comme pour les pipettes en verre, il est important, dans le cas des pipettes automatiques, qu'un liquide qui est pipeté soit similaire à celui avec lequel la pipette a été calibrée. Tant que nous pipetons des solutions aqueuses diluées, tout va bien. Mais si la pipette est calibrée avec de l'eau comme c'est habituellement le cas, et si la solution qui est ensuite pipetée est une solution saline concentrée ou une solution alcaline ou acide concentrée, une incertitude supplémentaire assez importante peut apparaître. Cela peut venir en grande partie de la différence de densité des liquides, et la différence de densité fera que moins de liquide sera retenu dans cette pipette parce qu'un léger vide se formera ici, de sorte qu'une partie du liquide tombera avant que vous puissiez retirer la pipette du liquide. L'effet de la température a bien sûr la même influence que dans la pipette en verre, mais là encore, c'est plus délicat. Si la pipette est tenue en main pendant une longue période, la température de cette pièce mécanique augmente et elle peut être nettement plus chaude que la température ambiante. Ainsi, l'effet de température de ce type de pipette peut être plus important. Il existe également d'autres sources d'incertitude qui font plutôt référence à des pratiques de travail bâclées : l'une d'elle est liée à l'utilisation d'un mauvais embout, la suivante est d'utiliser le bon embout mais de ne pas l'attacher correctement, et puis, si le pipetage est fait avec un angle, cela introduit aussi une incertitude supplémentaire. Il est vrai que, dans certaines applications, il est beaucoup plus facile pour les gens de pipeter sous un certain angle plutôt que verticalement, et dans ce cas, les gens calibrent souvent la pipette aussi sous ce même angle, ce qui est alors correct. |
Les résultats de mesure comportent une incertitude car il existe des sources d’incertitude (effets générateurs d’incertitude). Ce sont des effets qui entraînent des écarts de la valeur mesurée par rapport à la valeur réelle. Si la procédure de mesure utilisée est bien connue, les sources d'incertitude les plus importantes sont généralement également connues. Des efforts devraient être fait pour minimiser et, si possible, éliminer les sources d'incertitude en optimisant la procédure de mesure (procédure d'analyse). Les sources d'incertitude qui ne peuvent pas être éliminées (et il n'est jamais possible d'éliminer toutes les sources d'incertitude) doivent être prise en compte dans l'estimation de l'incertitude.
L'ampleur des déviations causées par des sources d'incertitude est généralement inconnue et, dans de nombreux cas, ne peut pas être connue. Ainsi, ces déviations ne peuvent qu'être estimés. Si nous pouvons estimer l'ampleur de chacune des sources d'incertitude importantes, nous pouvons les combiner et obtenir une estimation de l'incertitude de mesure, qui dans ce cas sera appelée incertitude composée. La façon dont cette combinaison est mathématiquement réalisée sera démontrée dans les prochains chapitres.
Si nous faisons plusieurs mesures répétées du même mesurande, alors idéalement, toutes ces mesures répétées devraient donner exactement la même valeur et cette valeur devrait être égale à la valeur vraie du mesurande. En réalité, les résultats des mesures répétées diffèrent presque toujours dans une certaine mesure et leur valeur moyenne diffère également de la valeur réelle. Les sources d'incertitude sont la cause. De manière quelque peu simplifiée, les sources d'incertitude (ou effets) peuvent être divisées en effets aléatoires et effets systématiques. Le schéma suivant illustre ceci (les cercles verts indiquent les valeurs vraies, les cercles jaunes les valeurs mesurées):
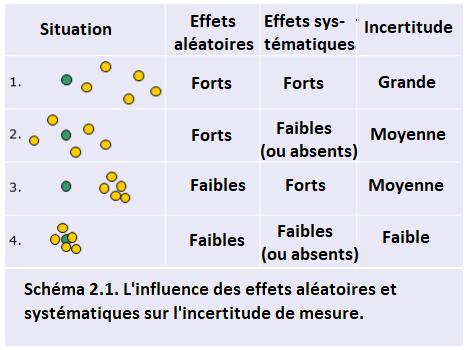
Les effets aléatoires provoquent une différence entre les résultats répétés de mesure (et donc, bien sûr, aussi avec la valeur vraie). Cependant, si un grand nombre de mesures répétées est effectué, la valeur moyenne sera peu influencée par les effets aléatoires (situation 2 sur le schéma). Ainsi, l’influence des effets aléatoires peut être diminuée en augmentant le nombre de répétitions. Les effets systématiques entraînent une déviation identique de toutes les mesures de la série dans la même direction. L'augmentation du nombre de répétitions ne permet pas de diminuer leur influence (situation 3 sur le schéma).
En principe, il est souhaitable de déterminer l’ampleur et la direction des effets systématiques et de corriger les résultats de mesure pour les effets systématiques. Cependant, cela peut souvent être très difficile et trop exigeant en terme de temps de travail, que cela devient impraticable. Par conséquent, dans de nombreux cas, plutôt que de déterminer exactement les effets systématiques et de les corriger, leur ampleur possible est estimée et est prise en compte comme source d’incertitude. Dans les chapitres 5.4 et 6, les effets aléatoires et systématiques sont traités de manière plus complète.
Il existe en général quatre sources principales d’incertitude dans les mesures volumétriques, à savoir les mesures à l’aide de pipettes, de burettes, de éprouvettes graduées et de fioles jaugées:
- Incertitude due à la répétabilité non idéale de la mesure (souvent appelée incertitude de répétabilité). Dans le cas du pipetage, cela signifie que si nous essayons avec soin de remplir et de vider la pipette, nous obtiendrons néanmoins à chaque fois un volume légèrement différent. Cela est parfois appelé «effet humain» ou «facteur humain», mais en fait, si une machine effectue le pipetage, il y aurait également une différence entre les volumes (bien que probablement plus petite). La répétabilité est un effet aléatoire typique et contribue à l'incertitude avec les pipettes en verre ainsi qu'avec les pipettes automatiques. Son influence sur le résultat de la mesure peut être diminuée en effectuant des mesures répétées, mais elle ne peut jamais être totalement éliminée.
- Incertitude due à l'étalonnage de l'équipement volumétrique (souvent appelée incertitude d'étalonnage). Dans le cas de la verrerie volumétrique, c’est l’incertitude sur la position des marques sur la verrerie volumétrique. Dans le cas des pipettes automatiques, cette incertitude est due au déplacement systématiquement trop élevé ou trop faible du piston à l'intérieur de la pipette. Dans le cas d'une pipette donnée, il s'agit d'une source d'incertitude systématique typique. Cette source d'incertitude peut être considérablement réduite en recalibrant la pipette dans le laboratoire par la personne qui travaille réellement avec elle. Le pesage exact de l’eau à température contrôlée est la base de l’étalonnage des instruments volumétriques, ainsi que la manière dont les incertitudes de répétabilité des différents instruments volumétriques sont généralement trouvées.
- Incertitude due à l'effet de la température (souvent appelée incertitude de la température). Tous les appareils volumétriques sont généralement calibrés à 20°C et les volumes se réfèrent généralement à des volumes à 20°C. La densité du liquide change (presque toujours diminue) avec la température. Si le pipetage est effectué à une température supérieure à 20°C, la quantité de liquide pipetée (en masse ou en nombre de molécules) est plus petite que si elle était effectuée à 20°C. Par conséquent, le volume de cette quantité de liquide à 20°C est également plus petit que si le pipetage était effectué à 20 C. Dans le cas de verreries volumétriques, la température affecte les dimensions de la vaisselle volumétrique (son volume augmente avec la température). Comme l'effet du changement de densité de liquide est environ 10 fois plus important, le changement de volume de la verrerie volumétrique est presque toujours négligé. Dans le cas des pipettes automatiques, l’effet de la température est plus complexe. Si l'air à l'intérieur de la pipette se réchauffe, le volume de liquide distribué peut varier dans une certaine mesure. Si la température du laboratoire et, ce qui est important, la température du liquide à la pipette, est constante pendant les mesures répétées, l'effet de la température est un effet systématique.
- Sources d'incertitude spécifiques à l'application. Celles-ci ne sont pas causées par l'équipement volumétrique, mais par le liquide manipulé ou par le système faisant l'objet de l'enquête. Quelques exemples:
1. Si une solution moussante est pipetée, mesurée au moyen d’une fiole jaugée ou d’une éprouvette graduée, on ne sait pas où se situe exactement la «solution», c’est-à-dire qu’il n’y a pas de ménisque bien défini. Cela provoquera une incertitude supplémentaire. Selon la situation, cet effet peut être aléatoire ou systématique ou inclure une partie aléatoire et une partie systématique.
2. Si le titrage est effectué à l'aide d'un indicateur visuel, le point final du titrage, c'est-à-dire le moment où l'indicateur change de couleur, est supposé correspondre au point d'équivalence (le point de stœchiométrie). Toutefois, en fonction de la réaction de titrage et de l'analyte titré, le résultat final peut être obtenu plus tôt ou plus tard que le point d'équivalence. Dans le cas du titrage donné, il s'agira d'un effet systématique. Cet effet peut être minimisé par un autre moyen de détection de point final, par ex. titrage potentiométrique
Il existe certaines autres sources d'incertitude qui s'avèrent généralement moins importantes, car elles peuvent être minimisées ou éliminées par des pratiques de travail correctes (toutefois, elles pourraient être importantes si ces pratiques n'étaient pas correctement appliquées). Les effets restants affectent généralement la répétabilité du pipetage ou son incertitude d'étalonnage et peuvent être pris en compte dans ces sources d'incertitude.
- Si la pipette n'est pas conservée verticalement (pipettes en verre et automatiques), l'attente n'est pas assez longue après la fin du drainage de la solution (pipette en verre), le volume de la pipette sera alors inférieur à celui obtenu avec un pipetage correct. Aucune attente n'est nécessaire dans le cas des pipettes automatiques car il ne reste pas de film liquide (et ne doit pas rester) sur la pipette jusqu'aux parois internes.
- Lors de l'utilisation d'une pipette en verre, il est toujours possible que de petits résidus de la solution précédente restent dans la pipette. Il est donc judicieux de rincer la pipette avant de pipeter (par exemple deux fois) avec la solution à pipeter (et de la jeter avec la solution de rinçage dans les déchets, pas dans le récipient d'où la solution a été prélevée). Dans le cas des pipettes automatiques, il est judicieux d’utiliser un nouvel embout chaque fois qu’une nouvelle solution est pipetée. Dans ce cas, cette contamination est généralement négligeable. En outre, lorsque vous pipetez plusieurs fois la même solution avec la même pipette, il est judicieux de contrôler l'absence de gouttelettes sur les parois internes et de remplacer la pointe lorsque les gouttelettes apparaissent.
- Si les parois d'une pipette en verre ne sont pas propres, des gouttelettes peuvent rester sur les parois après le drainage de la solution de pipetage. Cela conduit à un volume différent du cas où il ne reste plus de gouttelettes sur les parois de la pipette après avoir drainé la solution. La chose évidente à faire est de nettoyer la pipette.
- Si le liquide pipeté est très différent de l'eau (par exemple, un liquide très visqueux, tel que de l'huile végétale), le volume pipeté peut être systématiquement différent du volume nominal de la pipette. Cet effet existe à la fois avec les pipettes en verre et avec les pipettes automatiques. Dans un tel cas, la pipette doit être recalibrée avec le liquide en question ou la pesée doit être utilisée à la place de la volumétrie
Dans le chapitre 4, les sources d'incertitude du pipetage (la même expérience de pipetage que celle présentée dans la vidéo) seront quantifiées et combinées dans l'estimation de l'incertitude de mesure du volume pipeté. Les chapitres 4.1 à 4.5 présentent le calcul de l'incertitude à l'aide d'une pipette étalonnée en usine. Le chapitre 4.6 présente un exemple de calcul d’incertitude de mesure du volume de pipette à l’aide d’une pipette auto-étalonnée. Le chapitre 5 donne un aperçu de la majorité des sources d'incertitude rencontrées lors d'une analyse chimique.
Autoévaluation sur cette partie du cours : Test 2
4. Les concepts et outils fondamentaux
https://sisu.ut.ee/measurement/distribution-functions-normal-gaussian-distribution
Résumé : Cette section présente les concepts et outils les plus fondamentaux pour une estimation pratique de l’incertitude de mesure. Premièrement, les concepts de quantités aléatoires et de fonctions de distribution sont présentés. Ensuite, la distribution normale - la fonction de distribution la plus importante dans la science de la mesure - est expliquée et ses deux paramètres principaux - la valeur moyenne et l'écart type - sont introduits (3.1). Sur la base de l'écart type, le concept d'incertitude type est introduit (3.1, 3.2). Ensuite, l'estimation de l'incertitude de type A et de type B sont introduites (3.3). La valeur moyenne des quantités aléatoires est également une quantité aléatoire et sa fiabilité peut être décrite par l'écart type de la moyenne (3.4). Outre la distribution normale, trois fonctions de distribution supplémentaires sont introduites: la distribution rectangulaire et triangulaire (3.5) ainsi que la distribution de Student (3.6).
4.1. La fonction normale de distribution
https://sisu.ut.ee/measurement/31-normal-distribution
Résumé : Ce cours commence par généraliser que toutes les valeurs mesurées sont des quantités aléatoires du point de vue de la statistique mathématique. La distribution la plus importante en science de la mesure - la distribution normale - est ensuite expliquée : son importance, les paramètres de la distribution normale (moyenne et écart type). Les définitions initiales d'incertitude type (u), d'incertitude élargie (U) et de facteur d'élargissement (k) sont données. Un lien entre ces concepts et la distribution normale est créé.
La distribution normale
http://www.uttv.ee/naita?id=17589
| From the point of view of mathematical statistics, our measured quantities are actually random quantities. And random quantities are described by distribution functions. And when we deal with measurement science, then the most important distribution function for us is the normal or the Gaussian distribution. And because of certain laws of nature, whenever a process or a result is influenced by many factors, then the more factors we have that influence, the nearer the distribution function of that result becomes to the normal distribution. Therefore, very often measurement results are distributed normally, and the large part of the mathematics that is used in measurement science is based on the properties of the normal distribution. The normal distribution function curve is depicted in this slide. It characterizes the probability of a random quantity, and the random quantity is Y in our case. And the probability is on the vertical axis. And this distribution function tells us that there's a certain most probable value, the mean value Yn, where the probability of this random quantity occurring is highest. And as we depart from the mean value to either of these two sides, the probability gradually decreases. So that this is basically a probability density graph; the highest probabilities in the middle, the lowest on the sides. There are some important properties of this distribution function. First of all, even though the probability gradually decreases and actually quite sharply decreases as we move away from the main value, it never really becomes a zero. So there will also always be some positive nonzero probability. And this is the main reason why usually we cannot give measurement uncertainties 100% probability that we saw in another lecture. This normal distribution function property actually tells us that this is impossible. Now the second important feature this normal distribution curve is that even though it never really ends. so there always will be positive value however far we move from the mean value. Nevertheless, the area, the area under this curve is finite, and in the case of normalized function, it's equal to unity. Meaning the probability of this value being somewhere is 100%, which obviously needs to be true. Now let us examine the normal distribution function work closely on the example of pipetting. So we have a 10 mL pipette and suppose we pipette several times, trying to pipette ten milliliters every time. What happens? What happens is that every time, we get a slightly different value. So, see, it's always somewhere near ten milliliters, but never exactly ten. And also, it tends to be quite different, different. If we now plot these values as a histogram, then, we see something like this. This histogram in principle should resemble the beautiful bell-shaped curve that we just saw. But we see that it is, in fact, very different. The reason is obvious: we have only made ten measurements up to now, and ten measurements is a too small number to get the normal distribution curve. Let us also look at this axis here, it's now not probability, but frequency. Probability and frequency are closely connected to each other. Basically, the higher the probability, the higher the frequency of a certain measurement result occurring within a certain volume. Let us examine now what happens if we make more measurements. So as the measurements come, we see that they tend to come into the center part more often than on the sides. We can see that if we now make 27 measurments, then even though we still don't have this nice bell-shaped curve we already now have more measurement results in the middle and less measurement results on the sides. So that the number of measurement is increasing and this distribution function depicted here by histogram becomes more normal distribution like. But if we now make many a very large number of measurements, let's say 1,000 then our histogram really will resemble the normal distribution very nice. It is not really easy to make 1,000 measurements whatever the measurement is. But luckily we do not need nearly as many measurements to get the important characteristics of the normal distribution. So usually we do not need to record this shape as such. We simply need to know some important characteristics or important parameters. And those most important characteristics of parameters are two. The mean value, which characterizes the position of this curve, and the standard deviation S which characterizes its width or the scatter of the results. So the smaller the standard deviation, the smaller the scatter of the results. And now, with the use of this curve, we can make some very important observations about its nature. So we can see that if we move from the mean value away by one standard deviation to either of the sides, then we get a contour of this shape. I turns out, from the properties of the normal distribution function, that the ratio of this area to the overall area under this normal distribution curve, is a roughly 68%. This is the basis of definition of standard uncertainty. So standard uncertainty, as we see in many cases in other lectures, is the uncertainty given at one standard deviation level and also it's there by uncertainty at roughly 68% probability. Now we can multiply this standard deviation by some factor and if we multiply it by two, then we get this kind of curve. And now the area of this part here as related to the whole area under the normal distribution curve makes up roughly ninety five point five percent. And this here forms the basis of the so called expanded uncertainty. So expanded uncertainty, as we can see in other lectures, is obtained by multiplying standard uncertainty with some coverage factor. Two in this case. And it allows us, enables us obtaining measurement uncertainty estimate with higher probability. So this 2's level or as we say expanded uncertainty at k2 level, is uncertainty at roughly 95,5% probability. So the probability of the true value falling within this range is now significantly high. And likewise, if we multiply S by 3 then the same percentage will be 99.7% | Du point de vue des statistiques mathématiques, les quantités que nous mesurons sont en fait des quantités aléatoires. Les quantités aléatoires sont décrites par des fonctions de distribution. Quand nous avons affaire à la science de la mesure, alors la fonction de distribution la plus importante pour nous est la loi normale ou la distribution Gaussienne. À cause de certaines lois naturelles, lorsqu'un procédé ou un résultat est influencé par plusieurs facteurs, alors, plus on a de facteurs qui ont une influence, plus la fonction de distribution du résultat tend vers une distribution normale. Par conséquent, les résultats de mesure sont très souvent distribués suivant la loi normale. La plupart des lois mathématiques qui sont utilisées dans la science de la mesure sont basées sur des propriétés de la distribution normale. La courbe de la fonction de distribution normale est illustrée dans cette diapositive. Elle caractérise la probabilité d'une quantité aléatoire, qui est dans notre cas Y. La probabilité est sur l'axe vertical. Cette fonction de distribution nous dit qu'il existe une certaine valeur, la plus probable, la valeur moyenne Yn, où la probabilité que cette quantité aléatoire se produise est maximale. À mesure que nous nous éloignons de la valeur moyenne de part et d'autre, la probabilité diminue progressivement. Il s'agit donc d'un graphique de densité de probabilité avec au milieu, les plus grandes probabilités et les plus faibles sur les côtés. Il existe des propriétés importantes de cette fonction de distribution. Premièrement, même si la probabilité diminue progressivement et plutôt rapidement en s'écartant de la valeur moyenne, elle n'est jamais égale à zéro. Donc il y aura toujours des valeurs de probabilités positives et non nulles. C'est la raison pour laquelle on ne peut pas donner des incertitudes de mesures avec une probabilité de 100% comme nous l'avons vu dans d'autres cours. Cette propriété de la fonction de distribution normale nous dit donc que cela est impossible. La deuxième caractéristique importante de cette courbe de distribution normale est que même si elle ne se termine jamais, il y a donc toujours une valeur positive peu importe à quel point on s'éloigne de la valeur moyenne. Toutefois, l'aire sous la courbe reste finie. Dans le cas de la fonction normale elle est égale à 1. Ce qui signifie que la probabilité de cette valeur d'être quelque part est de 100%, ce qui est, évidemment, un fait. Maintenant, examinons de plus près la fonction de distribution normale qui s'applique ici à l'exemple du pipettage. Nous avons donc une pipette de 10 mL et supposons que nous avons voulu pipetter plusieurs fois en essayant de pipetter 10mL à chaque fois. Que se passe-t-il ? Ce qui se passe, c'est qu'à chaque fois on a une valeur légèrement différente. Nous pouvons voir que c'est toujours autour de 10mL mais jamais exactement 10. Cela a tendance à être différent. Si maintenant, nous relevons les données et que nous en faisons un histogramme, nous observons quelque chose comme ça. Cet histogramme devrait en principe ressembler à la belle courbe en forme de cloche que l'on vient de voir. Mais on constate que c'est très différent. La raison est évidente, nous avons seulement fait 10 mesures jusqu'à maintenant et c'est un nombre trop petit pour avoir une courbe de distribution normale. Regardons aussi cet axe (vertical), il ne s'agit plus de probabilités mais de fréquences. Probabilités et fréquences sont étroitement connectées car plus la probabilité est grande, plus la fréquence d'une certaine mesure de volume est élevée. Intéressons nous maintenant à ce qu'il se produit lorsque nous effectuons plus de mesures. Au fil des mesures, les valeurs de volume tendent plus souvent vers la partie centrale que vers les côtés. Si nous réalisons à présent 27 mesures, même si nous n'obtenons toujours pas la courbe en cloche, nous avons quand même plus de mesures au centre que sur les côtés. Donc, le nombre de mesures augmente et la fonction de distribution décrite ici par l'histogramme se rapproche d'une distribution normale. Si nous faisons un nombre encore plus élevé de mesures, disons 1000, alors notre histogramme ressemblera vraiment à une distribution normale. Ce n'est pas vraiment simple de faire 1000 mesures, peu importe la mesure dont il s'agit. Heureusement, nous n'avons pas besoin d'autant de mesures pour constater les caractéristiques de la distribution normale. Habituellement, nous n'avons pas besoin d'obtenir cette courbe (gaussienne). Nous avons juste besoin de connaitre les caractéristiques importantes ou les paramètres importants. Ces derniers sont au nombre de deux. La valeur moyenne qui caractérise la position de cette courbe, et l'écart type, S ,qui caractérise sa largeur ou la dispersion des résultats. Plus l'écart type est petit, moins la dispersion est grande. Grâce à l'étude de cette courbe, nous pouvons effectuer des remarques très importantes à propos de sa nature. Si nous nous éloignons de la valeur moyenne d'une unité d'écart type, de part et d'autre, alors nous avons le contour d'une partie de la courbe. Et il s'avère qu'avec les propriétés de la distribution normale, le ratio de l'aire de cette forme par rapport à l'aire totale sous la courbe est d'environ 68 %. Ceci est la base de la défintion de l'incertitude type (u). Cette dernière, comme nous l'avons vu dans plusieurs cas dans d'autres cours, est l'incertitude donnée pour une unité d'écart type et est d'environ 68%. Nous pouvons multiplier l'écart type par des facteurs et si nous le mutliplions par deux alors nous obtenons ce type de courbe. Et maintenant, le ratio de l'aire de cette courbe par rapport à l'aire totale sous la courbe de distribution normale correspond à environ 95,5%. Et cela forme la base de ce que l'on appelle l'incertitude élargie (U). Cette dernière, comme indiqué dans d'autres cours, est obtenue en multipliant l'incertitude type avec avec des facteurs d'élargissement (k). Deux dans ce cas. Et cela nous permet d'obtenir l'incertitude de mesure obtenue avec le plus de probabilité. Ce niveau "2s" ou plutôt incertitude élargie à un niveau de facteur 2, est l'incertitude à environ 95,5% de probabilité. Donc, la probabilité que la valeur vraie tombe dans cet intervalle, est significativement élevée. Pareillement, si l'on multiplie S par 3, alors ce pourcentage sera de 99,7%. |
Toutes les quantités mesurées (mesurandes) sont du point de vue des statistiques mathématiques des quantités aléatoires. Les quantités aléatoires peuvent avoir différentes valeurs. Cela a été démontré dans la vidéo sur l'exemple du pipettage. Si le pipettage avec une même pipette de volume nominal 10 ml est répété plusieurs fois, tous les volumes de pipettage tournent autour de 10 ml, mais restent légèrement différents. Si un nombre suffisamment grand de mesures répétées est effectué et si les volumes [1] pipettés sont tracés en fonction de leur fréquence d'utilisation, il devient évident que bien que aléatoires, les valeurs restent régies par une relation sous-jacente entre le volume et la fréquence : la probabilité maximale d'un volume est comprise entre 10,005 et 10,007 ml et sa probabilité décroît progressivement vers des volumes de plus en plus grands. Cette relation s'appelle une fonction de distribution.
Les mathématiciens connaissent de nombreuses fonctions de distribution et la plupart d’entre elles se rencontrent dans la nature, c’est-à-dire qu’elles décrivent certains processus de la nature. En science de la mesure, la fonction de distribution la plus importante est la distribution normale (également appelée distribution gaussienne). Son importance découle du soi-disant théorème de la limite centrale. De manière simplifiée, les mesures peuvent être libellées comme suit : si un résultat de mesure est simultanément influencé par de nombreuses sources d’incertitude, si le nombre des sources d’incertitude approche de l’infini, la fonction de distribution du résultat de mesure se rapproche de la distribution normale, quel que soit le résultat et les fonctions de distribution des facteurs / paramètres décrivant les sources d'incertitude. En réalité, la fonction de distribution du résultat devient déjà impossible à distinguer de la distribution normale s'il existe 3 à 5 sources (en fonction de la situation) contribuant de manière significative à l'incertitude [2]. Cela explique pourquoi, dans de nombreux cas, les quantités mesurées ont une distribution normale et pourquoi l'essentiel de la base mathématique de la science de la mesure et de l'estimation de l'incertitude de mesure est basée sur la distribution normale.
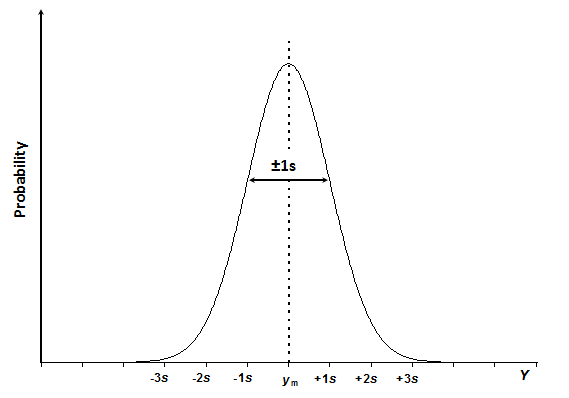
Schéma 3.1. La courbe de distribution normale de la quantité Y avec la valeur moyenne ym et l'écart type s.
La courbe de distribution normale a l'aspect d'une cloche (schéma 3.1) et est exprimée par l'équation 3.1:
 (3.1)
(3.1)
Dans cette équation, f( y ) est la probabilité que le mesurande Y ait la valeur y. ym est la valeur moyenne de la population et s, l'écart type de la population. ym caractérise la position de la distribution normale sur l'axe des ordonnées, s caractérise la largeur (étendue) de la fonction de distribution, qui est déterminée par la dispersion des points de données. La moyenne et l'écart type sont les deux paramètres qui déterminent entièrement la forme de la courbe de distribution normale d'une quantité aléatoire donnée. Les constantes 2 et pi sont des facteurs de normalisation, qui sont présents afin de rendre l’aire totale sous la courbe égale à 1.
Le mot «population» signifie ici que nous aurions besoin de réaliser un nombre infini de mesures afin d’obtenir les vraies valeurs ym et s. En réalité, nous travaillons toujours avec un nombre limité de mesures, de sorte que la valeur moyenne et l'écart type que nous avons dans nos expériences sont en fait des estimations de la moyenne vraie et de l'écart type réel. Plus le nombre de mesures répétées est grand, plus les estimations sont fiables. Le nombre de mesures parallèles est donc très important et nous y reviendrons dans différentes parties de ce cours.
La distribution normale et l'écart type constituent la base de la définition de l'incertitude type. L'incertitude type, appelée u, est l'incertitude exprimée au niveau de l'écart type, c'est-à-dire une incertitude avec une probabilité de couverture d'environ 68,3% (c'est-à-dire que la probabilité que la valeur vraie se situe dans la plage d'incertitude est d'environ 68,3%). La probabilité de 68,3% est souvent trop faible pour des applications pratiques. Par conséquent, l’incertitude des résultats de mesure n’est dans la plupart des cas pas rapportée en tant qu’incertitude type, mais en tant qu’incertitude élargie. L'incertitude élargie, notée U, est obtenue en multipliant l'incertitude type par un facteur d'élargissement [3], noté k, qui est un nombre positif supérieur à 1. Si le facteur d'élargissement est par exemple égal à 2 (qui est la valeur la plus utilisée pour le facteur d'élargissement), dans le cas d'un résultat de mesure normalement distribué, la probabilité de couverture est d'environ 95,5%. Ces probabilités peuvent être considérées comme des fractions d'aire des segments respectifs de l'aire totale sous la courbe, comme illustré par le schéma suivant.
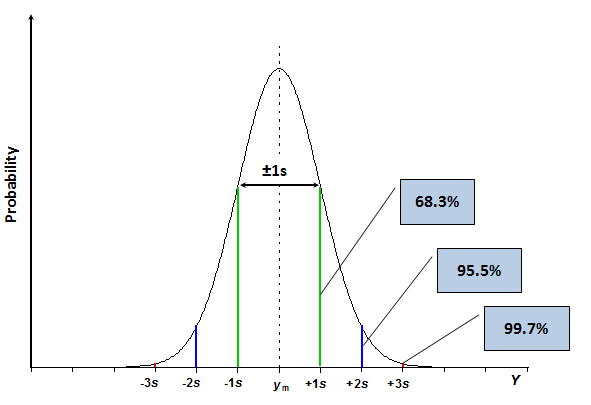
Schéma 3.2. La même courbe de distribution normale que dans le schéma 3.1 avec les segments 2 et 3 indiqués.
Puisque la fonction exposant ne peut jamais renvoyer la valeur zéro, la valeur de f( y ) dans l'équation 3.1 est supérieure à zéro quelle que soit la valeur de y. C’est la raison pour laquelle l’incertitude avec une couverture de 100% n’est (presque) jamais possible.
Il est important de souligner que ces pourcentages ne sont valables que si le résultat de la mesure est normalement distribué. Comme dit plus haut, c'est très souvent le cas. Il existe cependant des cas importants où le résultat de la mesure n'est pas normalement distribué. Dans la plupart des cas, la fonction de distribution a des «queues plus lourdes», ce qui signifie que l’incertitude accrue, par exemple le niveau k = 2, ne correspondra pas à une probabilité de couverture de 95,5%, mais à une probabilité inférieure (par exemple 92%). La question de la distribution du résultat de la mesure dans ce cas sera abordée plus tard dans ce cours.
Autoévaluation sur cette partie du cours : Test 3.1
https://sisu.ut.ee/measurement/node/1396
***
[1] Il est légitime de se demander : comment pouvons nous connaître les volumes prélevés individuellement si la pipette nous “dit” seulement que le volume est de 10mL ? En fait, nous avons seulement la pipette (plus juste) et aucune autre possibilité de mesure du volume, donc on ne peut pas savoir de combien les volumes diffèrent les uns des autres ou par rapport à la valeur nominale. Cependant, si une méthode plus exacte est disponible, alors cela devient possible. Dans le cas du pipettage, une méthode très adaptée et très utilisée est la pesée. Il est possible de trouver le volume d’eau pipetté, qui est plus juste que le volume obtenu juste en pipettant, en pesant la solution prélevée (le plus souvent l’eau) et en divisant la masse obtenue par la densité de l’eau à la température de celle-ci. L’eau est utilisée dans de telles expériences car les densités de l’eau à différentes température sont bien connues (voir : https://en.wikipedia.org/wiki/Properties_of_water#Density_of_water_and_ice).
[2] Les sources d’incertitude dont la contribution est significative sont celles qui sont importantes. Nous avons déjà vu de manière qualitative dans la section 2 que les différentes sources d’incertitude n’ont pas la même importance. Dans les cours à suivre nous verrons également comment “l’importance” d’une source d’incertitude (sa contribution à l’incertitude) peut être exprimée de manière quantitative.
[3] Cette définition de l’incertitude élargie est simplifiée. Une définition plus rigoureuse existe via l’incertitude-type composée sera introduite en section 4.4.
4.2. Moyenne, écart type, incertitude de mesure
https://sisu.ut.ee/measurement/32-mean-standard-deviation-and-standard-uncertainty
Résumé : Ce cours explique le calcul de la moyenne (Vm) et de l’écart type (s), illustrant encore la probabilité de 68% associée à s. Il explique comment l’incertitude type de la répétabilité u (V, REP) peut être estimée en tant qu’écart type des résultats de mesure en parallèle. Il souligne l’importance de l’incertitude type en tant que paramètre clé dans la réalisation des calculs d’incertitude : les incertitudes correspondant à différentes sources (pas seulement la répétabilité) et à différentes fonctions de distribution sont converties en incertitudes-types lorsque des calculs d’incertitude sont effectués.
Moyenne, écart type, incertitude de mesure
http://www.uttv.ee/naita?id=17554
| Let us now take a closer look at the mean value and standard deviation. And we will do that again on the example and we will use pipetting example again for this. So suppose we have a pipette and we are pipetting a volume which we denote by VG. And we pipetting at several times and we get each time a slightly different volume because of the random effects that affect pipetting. So the volumes that we actually get are the following : V1 V2 V3 etc until Vn so these are here the individual volumes and n is the number of measurements. Now with these volumes , with these values on volume obtained during those parallel pipetting operations we can calculate both the mean and the standard deviation very easily. We denote mean by Vm and it is obviously found by summing up all those individual volumes and dividing by the number of the volumes. And this thing can be equally well written as follows. So here we denote by the Sigma sign the sum of all these volumes, each and every of those individual volumes here is denoted by the single Vi and i changes from 1 to n so that these two equations are equivalent. And now the standard deviation is calculated as follows. Here it is. so it is the square root of this term which in the numerator has again a sum again from 1 to n meaning involving all those volumes but now we sum up the squares of differences of each and every of this volume and the mean volume and we divide all that by n minus 1 and finally take the square root. So mathematically speaking, standard deviation is something like root mean square of differences of the individual volumes from the mean volume and this minus 1 comes from the fact that we do not do infinite number of measurements but we do a finit number of measurement. Now, the mean value for us characterizes the most probable volume that we obtain when we make pipeting. So every next volume that we're pipeting using the same pipet under these same conditions will most probably be very near to the mean value. But standard deviation characterizes the scatter, the spread of those values. It characterizes how well our parallel pipetting operations agree between themselves and obviously the smaller is the standard deviation the better we are in pipeting. So now, this standard deviation can be interpreted also in two very important ways. So first of all if now we have calculated this mean value and the standard deviation, then the probability of every next pipeting falling within the mean plus minus one standard deviation region is roughly 68% and this percentage comes from the properties of the normal distribution function. So this is the first important thing and the second important thing we now come to the quantifying measurement uncertainty. And standard deviations are very important means, actually the most important means, of characterizing measurement uncertainty and in fact the uncertainty due to repeatability, meaning, due to random effects of any individual pipetted volume is characterized by standard deviation and this uncertainty is denoted as the standard uncertainty. So we can speak about the standard uncertainty, denoted by small u, of this volume which we measure by pipetting ; due to repeatability meaning this is one uncertainty components : the repeatability uncertainty component ; and this is equal to this same standard deviation. Let us now briefly summarize what we just saw. So the mean value characterizes the most probable value with that we can obtain by next pipeting. Then the standard deviation characterizes the scatter and also the standard deviation carries two important bits of information. On one hand it defines the range within which the probability of finding the next pipetted volume is roughly 68% ; and finally, via standard deviation, we define standard uncertainty which is denoted by small u and oftentimes is also called uncertainty at the standard deviation level or in other words, uncertainty at roughly 68% probability. In the very introductory slide, I explained that uncertainty range always involves a certain probability meaning we cannot define the range with 100% probability. So in this case, if uncertainty is expressed at the standard uncertainty level, the probability is roughly 68 %. And we will see later on that standard uncertainty is perhaps the most important way of presenting uncertainty for us because the calculations in uncertainty estimation all go via standard uncertainties, meaning if the uncertainty component of certain uncertainty sources are characterized by some other way they all will need to be transformed into standard uncertainties before they can be combined. | Examinons maintenant de plus près la valeur moyenne et l'écart-type. Et nous le ferons à nouveau sur un exemple et nous utiliserons encore l'exemple de pipettage pour cela. Donc supposons que nous avons une pipette et que nous pipettons un volume que nous notons VG. Et nous pipettons à plusieurs reprises et nous obtenons à chaque fois un volume légèrement différent en raison des effets aléatoires qui affectent le pipettage. Donc les volumes que nous obtenons réellement sont les suivants : V1, V2, V3 etc jusqu'à Vn. Donc ce sont ici les volumes individuels et n est le nombre de mesures. Maintenant avec ces volumes, avec ces valeurs de volume obtenus pendant ces opérations de pipettage en parallèles, nous pouvons calculer à la fois la moyenne et l'écart type très facilement. On note Vm la moyenne et on la trouve évidemment en faisant la somme de tous les volumes individuels et en divisant par le nombre de volume. Et cette chose peut être aussi bien écrite comme ceci. Donc, ici, on note par le symbole sigma la somme de tous les volumes. Chacun de ces volumes individuels ici est noté seulement sous le signe Vi et i change de 1 à n de sorte que ces deux équations soient équivalentes. Et maintenant l'écart-type est calculé comme ceci. Et voilà. C'est donc la racine carrée de ce terme qui, au numérateur, a à nouveau une somme de 1 à n signifiant l'implication de tous ces volumes mais maintenant nous faisons la somme des carrés des différences de chacun de ces volumes au volume moyen, et nous divisons l'ensemble par n moins 1 et on en fait finalement la racine carrée. Donc mathématiquement parlant, l'écart type est quelque chose comme la moyenne quadratique des différences des volumes individuels par rapport au volume moyen. Et ce "moins 1" vient du fait que nous ne faisons pas un nombre infini de mesures mais nous faisons un nombre fini de mesures. Maintenant, la valeur moyenne pour nous caractérise le volume le plus probable que nous obtenons lorsque nous pipettons. Donc, chaque volume suivant que nous pipettons en utilisant la même pipette, dans les mêmes conditions, sera très probablement très proche de la valeur moyenne. Mais l'écart type caractérise la dispersion, la propagation de ces valeurs. Il caractérise à quel point, nos opérations de pipettage parallèles s'accordent entre elles. Et évidemment, plus l'écart type est petit, meilleur est notre pipettage. Donc maintenant, cet écart type peut être également interprété de deux manières très importantes. Donc tout d'abord, si maintenant nous avons calculé cette valeur moyenne et l'écart type, alors la probabilité que chaque pipettage suivant tombe dans la gamme, moyenne plus ou moins un écart type est d'environ 68%. Ce pourcentage provient de la probabilité de la fonction normale de distribution. C'est donc la première chose importante et la seconde chose importante; nous arrivons maintenant à la quantification de l'incertitude de mesure. Et les écarts-types sont des moyens très important, en fait ,les moyens les plus importants, pour caractériser l'incertitude de mesure. Et en fait, l'incertitude due à la répétabilité, c’est-à-dire, due aux effets aléatoires de volume pipetté individuellement, est caractérisée par l'écart-type et cette incertitude est notée incertitude type. Donc nous pouvons parler de l'incertitude type, notée petit u, de ce volume que nous mesurons par pipettage dûe à la répétabilité, signifiant que c'est une composante d'incertitude; la composante d'incertitude de répétabilité et c'est égal à ce même écart-type. Résumons maintenant brièvement ce que nous venons juste de voir. Donc la valeur moyenne caractérise la valeur la plus probable que nous pouvons obtenir avec le pipettage suivant. Puis l'écart-type caractérise la dispersion .Et aussi l'écart-type comporte deux informations importantes. D'une part, il définit la gamme dans laquelle la probabilité de trouver le prochain volume pipetté est d'environ 68% ; et finalement, via l'écart-type, nous définissons l'incertitude type qui est notée avec un petit u et qui est aussi souvent appelée incertitude au niveau de l'écart-type. Ou dans d'autres mots, incertitude à une probabilité d'environ 68%. Dans la diapositive d'introduction, j'ai expliqué que la gamme d'incertitude implique toujours une certaine probabilité. Ce qui signifie que nous ne pouvons pas définir la gamme avec 100% de probabilité. Donc, dans ce cas, si l'incertitude est exprimée au niveau de l'incertitude type, la probabilité est d'environ 68%. Et nous verrons plus tard que l'incertitude type est peut-être la façon la plus importante de présenter l'incertitude pour nous car, les calculs dans l'estimation de l'incertitude passent tous par les incertitudes-types, ce qui signifie que si les composantes d'incertitude de certaines sources d'incertitude sont caractérisées d'une autre façon, elles devront toutes être transformées en incertitudes-types avant de pouvoir être combinées. |
L'une des approches les plus courantes pour améliorer la fiabilité des mesures consiste à répliquer des mesures de la même quantité. Dans un tel cas, très souvent, le résultat de la mesure est présenté comme la valeur moyenne des mesures répétées. Dans le cas de pipettage, on obtient n fois avec les mêmes volumes de pipette V1, V2,…, Vn et la valeur moyenne Vm est calculée comme suit:
 (3.2)
(3.2)
Comme expliqué à la section 3.1, la moyenne calculée de cette manière est une estimation de la valeur moyenne réelle (qui pourrait être obtenue s'il était possible d'effectuer un nombre infini de mesures).
La dispersion des valeurs obtenues à partir de mesures répétées est caractérisée par l'écart type des volumes de pipettage, qui, dans le même cas de pipettage, est calculé comme suit:
 (3.3)
(3.3)
Le n-1 du dénominateur est souvent appelé nombre de degrés de liberté. Nous verrons plus tard qu'il s'agit d'une caractéristique importante d'un ensemble ou de mesures répétées. Plus il est élevé, plus la moyenne et l'écart type peuvent être fiables.
Deux interprétations importantes de l'écart type:
- Si Vm et s(V) ont été trouvés grâce à un nombre suffisamment grand de mesures (généralement 10 à 15 suffisent), la probabilité que chaque mesure suivante (effectuée dans les mêmes conditions) tombe dans la plage Vm ± s (V) est environ 68,3%.
- Si nous effectuons plusieurs mesures répétées dans les mêmes conditions, l'écart-type des valeurs obtenues caractérise l'incertitude due à la répétabilité non idéale (souvent appelée incertitude type de répétabilité) de la mesure: u(V,REP)=s(V). La répétabilité non idéale est l’une des sources d’incertitude dans toutes les mesures.
L’écart-type est la base de la définition de l’incertitude type, ou incertitude au niveau de l’écart-type, indiquée par un petit u. Trois aspects importants de l’incertitude type méritent d’être soulignés ici:
- L'écart type peut également être calculé pour des quantités qui ne sont pas normalement distribuées. Cela permet d'obtenir pour eux des estimations d'incertitude type.
- De plus, les sources d’incertitude systématiques par nature et qui ne peuvent pas être évaluées par des mesures répétées peuvent toujours être exprimées numériquement sous forme d’estimations d’incertitude type.
- Convertir différents types d'estimations d'incertitude en incertitude type est très important car, comme nous le verrons à la section 4, la plupart des calculs de l'évaluation de l'incertitude, notamment en combinant les incertitudes correspondant à différentes sources d'incertitude, sont effectués à l'aide d'incertitudes-types.
L'incertitude-type d'une quantité (dans notre cas, le volume V) exprimée en unités de cette quantité est parfois appelée incertitude-type absolue. L'incertitude type d'une quantité divisée par la valeur de cette quantité est appelée incertitude type relative, urel (de la même manière que l'équation 1.1). Dans le cas du volume V:
![]() (3.4)
(3.4)
Autoévaluation sur cette partie du cours : Test 3.2
https://sisu.ut.ee/measurement/measurement-1-7
***
[1] Nous verrons plus loin que l'écart-type des mesures répétées dans des conditions qui changent de manière prédéfinie (c'est-à-dire qu'il ne s'agit pas de répétabilité) est également extrêmement utile dans le calcul de l'incertitude, car il permet de prendre en compte plusieurs sources d'incertitude simultanément.
4.3. Estimation des incertitudes, types A et B
https://sisu.ut.ee/measurement/33-and-b-type-uncertainty-estimates
Effectuer plusieurs fois la même opération de mesure et calculer l'écart-type des valeurs obtenues est l'une des pratiques les plus courantes en matière d'estimation de l'incertitude de mesure. La mesure complète, ou seulement certaines parties, peuvent être répétées. Dans les deux cas, des informations utiles peuvent être obtenues. L'écart-type obtenu (ou l'écart-type de la moyenne, expliqué à la section 3.4) est alors l'estimation de l'incertitude type.
Les estimations d'incertitude obtenues sous forme d'écarts-types de résultats de mesure répétés sont appelées estimations d'incertitude de type A.
Si l'incertitude est estimée en utilisant une autre méthode que le traitement statistique des résultats de mesures répétées, les estimations obtenues sont appelées estimations de l'incertitude de type B. Les autres moyens peuvent être, par exemple, certificats de matériaux de référence, spécifications ou manuels d'instruments, estimations basées sur une longue expérience, etc.
Estimation des incertitudes, types A et B
http://www.uttv.ee/naita?id=18165
| In very broad terms there are two ways of estimating measurement uncertainty components : the A type and the B type uncertainty estimates. And let us see what these terms mean. A-type uncertainty estimates are obtained from repeated the measurement results. So, whenever we repeat measurement several times and then make our calculations we do A type uncertainty estimation or uncertainty estimation of the type A. And usually, the data treatment means calculating the standard deviation. So the most common A type measurement uncertainty is a standard deviation. Secondly all such uncertainty estimates that happened without the use of repeated measurements are called B type or type B uncertainty estimates. And here we can bring as examples, for example, concentrations of standard solutions whereby the uncertainty of the concentration is obtained from the certificate in standard solution or some data from the instrument documentation or educated guesses or expert opinions, which in chemistry, are quite important. Those also qualify as B type uncertainty estimates. So, for example, a person working with a certain instrument during long long time can easily say that this instrument will not be off systematically by more than some magnitude. And this then would be called a B type uncertainty estimates. How do A type and B type uncertainty estimates relate to the random and systemic effects that we have looked ? There is a relation and there are some similarities between random effects and type A uncertainty estimation systemic effects and type B but these are not synonymous. Let us see. Random effects most usually are estimated by type A estimates. So all the repeatabilities within lab long-term reproducibilities and so on… are typical type A uncertainty estimates. Now, random effects can also sometimes evaluate in by type B estimation. And these are all those cases where we do not have the separate measurement data but we have either some summary characteristics or we get the data from some data source which does not provide to us the original data but only the eventual findings, the eventual uncertainty estimates. Let us look now at the systemic effect. Systemic effects typically are evaluated using type B evaluation or type B estimation. However, in certain cases, certain systemic effects can also be evaluated by type A uncertainty values. And these are all those cases when the effect is a systemic effect in the short term but in the long term can become a random effect In such a case type a evaluation is possible and this is what is typically done done in within lab long term reproducibility calculation which we see later on in this course. | En termes très généraux, il existe deux façons d'estimer les composantes de l'incertitude de mesure : les estimations d'incertitude de type A et de type B. Voyons ce que signifient ces termes. Les estimations d'une incertitude de type A sont obtenues à partir des résultats de mesure répétés. Ainsi, à chaque fois que nous répétons la mesure plusieurs fois, et que nous effectuons ensuite nos calculs, nous faisons une estimation d'incertitude type A ou une estimation de l'incertitude de type A. Et généralement, le traitement des données signifie calculer l’écart-type. Ainsi, l'incertitude de mesure de type A la plus courante est donc un écart-type. Deuxièmement, toutes ces estimations d'incertitude qui se sont produites sans utiliser de mesures répétées sont appelées de type B ou estimations de l'incertitude de type B. Et ici, nous pouvons apporter comme exemples, par exemple, des concentrations de solutions étalons par lesquelles l'incertitude de la concentration est obtenue à partir du certificat en solution étalon ou de certaines données de la documentation de l’instrument ou de déduction logique ou d'opinions d'experts qui, en chimie, sont assez importantes. Celles-ci sont également qualifiées d'estimations d'incertitude de type B. Ainsi, par exemple, une personne travaillant avec un certain instrument pendant très longtemps peut facilement dire que cet instrument ne sera pas éloigné systématiquement de plus d'une certaine ampleur. Et cela serait alors appelé une estimation de l'incertitude de type B. Comment les estimations d'incertitude de type A et de type B sont-elles liées aux effets aléatoires et systémiques que nous avons examinés ? Il existe une relation et il existe certaines similitudes entre les effets aléatoires et et les effets systémiques d'estimation de l'incertitude de type A et de type B, mais ceux-ci ne sont pas synonymes. Voyons, les effets aléatoires sont généralement estimés par des estimations de type A. Ainsi, toutes les répétabilités pour les études de reproductibilité sur le long terme au sein d'un laboratoire et ect... sont des estimations typiques de l’incertitude de type A. Maintenant, les effets aléatoires peuvent également parfois être évalués par une estimation de type B. Et ce sont tous ces cas où nous n'avons pas les données de mesure distinctes mais nous avons soit certaines caractéristiques résumées, soit nous obtenons les données d'une source de données qui ne nous fournit pas les données d'origine mais uniquement les conclusions éventuelles, les estimations de l'incertitude éventuelle. Voyons maintenant l'effet systémique. Les effets systémiques sont généralement évalués à l'aide d'une évaluation de type B ou d'une estimation de type B. Cependant, dans certains cas, certains effets systémiques peuvent également être évalués par des valeurs d'incertitude de type A. Et ce sont tous ces cas où l'effet est un effet systémique à court terme mais qui peut, à long terme, devenir un effet aléatoire. Dans un tel type de cas, une évaluation est possible et c'est ce qui est généralement fait dans le calcul de reproductibilité à long terme en laboratoire que nous verrons plus tard dans ce cours. |
4.4. Ecart type de la moyenne
https://sisu.ut.ee/measurement/33-standard-deviation-mean
Résumé : Comme les valeurs individuelles, la valeur moyenne calculée à partir de celles-ci est également une quantité aléatoire ce qui permet également de calculer un écart-type. Il est possible de calculer l'écart-type de la moyenne à partir de l'écart-type des valeurs individuelles. Il est expliqué ici quand utiliser l'écart-type de la valeur individuelle et quand utiliser l'écart-type de la moyenne : chaque fois que le résultat individuel est utilisé dans un calcul ultérieur, l'écart-type du résultat individuel doit être utilisé; chaque fois que la valeur moyenne est utilisée dans des calculs ultérieurs, l’écart-type de la moyenne doit être utilisé.
Ecart type de la moyenne
http://www.uttv.ee/naita?id= 17580
| Let us now take a look at the standard deviation. What we just saw, calculated according to this equation, is the standard deviation of the volume pipetted with the pipette and importantly we can also see that it is the standard deviation of the individual pipetting result. On the other hand now if we calculate the mean value, the mean value from the parallel pipetting measurements, then this mean value also will be random quantity it will also have a standard deviation and as we see later on very often this standard deviation of the mean will be very interesting for us. How to weaken it? On one hand, we could make a large number of pipetting series. Suppose we do fifteen pipetting operations, we calculate the mean and the standard deviation. We then do another fifteen pipetting, another, another, another fifteen pipetting operations so that we finally get a number of mean values and then we can simply calculate again the standard deviation from all those mean values. This is fully valid and fully doable. Unfortunately, it involves a very large number of measurements that usually is not practical in chemistry. Therefore, most of the time people use a very much simple approach to find the standard deviation of the mean. And it turns out that the standard deviation of the mean can be found from the standard deviation of the individual result simply by dividing by square root of n. Very easy. So we make just one series, large enough, say ten to fifteen measurements, find the standard deviation, divide it by the square root of n and here we have the standard deviation of the mean. It turns out now that we have two different standard deviations for the same series of measurement and it is very reasonable to ask when do we use the standard deviation of the individual result and when do we use the standard deviation of the mean. The general rule is the following : we use the one which corresponds to the actual value that we use. So, if we use for our purpose the individual value from this series, we use standard deviation of the individual value. On the other hand, if we use for our purpose the mean value then we also use the standard deviation of the mean value. | Abordons maintenant l'écart-type. Ce que nous venons de voir être calculé à partir de cette équation est l'écart-type du volume prélevé avec la pipette, et surtout, nous pouvons aussi voir que cela correspond à l'écart type du résultat de pipetage individuel. D’autre part, si on calcule la valeur moyenne, basée sur des mesures de pipetage parallèles, cette valeur moyenne sera alors elle-même une quantité aléatoire, elle aura aussi un écart-type et comme nous le verrons plus tard, l'écart-type de la moyenne sera souvent très intéressant pour nous. Comment le rendre le plus faible possible ? D'une part, nous pourrions réaliser un grand nombre de séries de pipetages. Supposons que nous réalisions quinze prélèvements à la pipette et que nous calculions la moyenne ainsi que l'écart-type. Nous faisons alors 15 prélèvements supplémentaire, puis encore et encore 15 prélèvements à la pipette afin d'obtenir un nombre de valeurs moyennes qui nous permettent de calculer simplement à nouveau l'écart-type à partir de ces valeurs moyennes. Cela est tout à fait correct et réalisable. Malheureusement, cela implique de réaliser de nombreuses mesures qui ne sont habituellement pas pratiques en chimie. Par conséquent, la plupart du temps on utilise une approche plus simple pour trouver l'écart-type de la moyenne. Il s'avère que l'écart-type de la moyenne peut être trouvé à partir de l'écart-type du résultat individuel simplement en le divisant par la racine carrée de n. C'est très simple. On ne réalise ainsi qu'une seule série, suffisamment importante, disons entre dix et quinze mesures, puis on trouve l'écart-type que l'on divise par la racine carrée de n et l'on obtient ainsi l'écart-type de la moyenne. Il s'avère que nous obtenons deux écarts-types différents pour la même série de mesures et il est tout à fait convenable de se demander quand utiliser l'écart-type du résultat individuel et quand utiliser l'écart-type de la moyenne. Voici la règle générale : il faut utiliser l'écart-type qui correspond à la valeur réelle que l'on utilise. De ce fait, si l'on utilise la valeur individuelle de la série, il faut utiliser l'écart-type de la valeur individuelle. D'autre part, si l'on utilise la valeur moyenne, il faut utiliser l'écart-type de la valeur moyenne. |
L'écart-type s(V) calculé à l'aide de la formule 3.3 est l'écart-type d'un résultat de pipetages individuels. Lorsque la valeur moyenne est calculée à partir d'un ensemble de valeurs individuelles réparties de manière aléatoire, la valeur moyenne sera également une quantité aléatoire. Comme pour toute quantité aléatoire, il est également possible de calculer l'écart-type pour la moyenne s(Vm). Une façon possible de le faire serait d'effectuer de nombreuses séries de mesures, de rechercher la moyenne pour chaque série, puis de calculer l'écart-type de toutes les valeurs moyennes obtenues. Ceci serait trop exigeant en terme de temps de travail. Il existe une approche beaucoup plus simple pour calculer s(Vm), il suffit de diviser s(V) par la racine carrée du nombre de mesures répétées effectuées :
 (3.5)
(3.5)
Ainsi, pour un ensemble de valeurs de pipetage répétés, nous avons en fait obtenu deux écarts types: l’écart-type de la valeur unique s(V) et l’écart-type de la moyenne s(Vm). Il est important de se demander quand on utilise l'un et quand on utilise l'autre ?
La règle générale est la suivante: lorsque la valeur mesurée indiquée ou utilisée dans les calculs ultérieurs est une valeur unique, nous utilisons l’écart-type de la valeur unique; quand il s'agit de la valeur moyenne, nous utilisons l'écart-type de la moyenne.
Illustrons ceci par deux exemples:
- Lorsque nous délivrons un certain volume à la pipette, le pipetage est une opération ponctuelle: nous ne pouvons pas répéter le pipetage avec la même quantité de liquide. Nous utilisons donc l’écart-type du pipetage simple comme incertitude de répétabilité du pipetage.
- Lorsque nous pesons une certaine quantité d'un matériau, nous pouvons le peser à plusieurs reprises. Donc, si nous devons minimiser l’influence de la répétabilité de pesée dans nos mesures, nous pouvons peser le matériau à plusieurs reprises et utiliser dans nos calculs la masse moyenne. Dans ce cas, l'écart-type de répétabilité de cette masse moyenne est l'écart-type de la moyenne. Si, par contre, il n’est pas très important d’avoir l’incertitude de masse la plus faible possible sur la répétabilité, nous pesons une seule fois et utilisons la valeur de masse de la pesée simple. En tant qu’incertitude de répétabilité, nous utilisons l’écart-type d’une valeur unique. [1]
Dans le cas d'un pipetage simple ou d'une pesée simple, l'incertitude de répétabilité ne peut évidemment pas être estimée à partir de cette opération unique. Dans ces cas, la répétabilité est déterminée séparément puis utilisée pour les mesures concrètes.
Autoévaluation sur cette partie du cours : Test 3.4
https://sisu.ut.ee/measurement/node/1405
***
[1] Comme nous le verrons plus tard, les balances modernes sont des instruments extrêmement fidèles et l'incertitude liée à la pesée fait rarement partie des sources importantes d'incertitude. Ainsi, à moins que certains effets perturbants ne gênent la pesée, il n’est généralement pas nécessaire de peser plusieurs fois les matériaux.
4.5. Distribution rectangulaire et triangulaire
Résumé : La distribution rectangulaire et la distribution triangulaire sont expliquées, ainsi que la manière dont les incertitudes correspondant à la distribution rectangulaire ou triangulaire peuvent être converties en incertitudes types. Souvent, les informations sur la fonction de distribution sont manquantes et ensuite, une fonction de distribution est généralement supposée ou postulée. Les distributions rectangulaires et triangulaires font partie des fonctions de distribution postulées les plus courantes. Des recommandations sont données.
Distribution rectangulaire et triangulaire
http://www.uttv.ee/naita?id=17584
| Let us now examine some other distribution functions besides the normal distribution. As I explained the normal distribution has unique importance in measurement science but there are a few others that are also important and we will now look at the rectangular and triangular distribution and we will do that on the example of a pipette again. We look at this pipette. The uncertainty of calibration of this pipette is 0.03 mL and if we look at it we see that there's a sign of plus minus in front of the 0.03 and there's no information given anywhere here about the status of this uncertainty about the distribution function or about the coverage factor. Let us see how we should handle this kind of information that comes in a limited way meaning there's a lack of concrete information about the uncertainty. The usual way that people handle this kind of situation is that some distribution function is assumed or is postulated and rectangular and triangular distributions are the most frequent ones to be used in this case. So let us look at the rectangular distribution. We have here the probability and possible values for the measurement and let it be the calibrated volume of the pipette. And this is here 10 mL. And now this uncertainty ±0.03 would be here like follows. And now if it is rectangular distribution then this means that the probability of the true calibrated volume being anywhere within this range is equal meaning that the area under this curve is unity. The probability of the volume being outside is zero and there's no distinction between any of these values here. Now if we want to use this uncertainty estimate this ±0.03 mL in us uncertainty calculation. We have to convert it somehow into standard uncertainty and converting from rectangular distribution to standard uncertainty is very easy and can be done as follows. It is simply divided by square root of three. So that whenever we have uncertainty expressed as plus/minus some value and we have a reason to assume rectangular distribution we always can find the standard uncertainty estimate by dividing by square root of three. Let us now look at the triangular distribution. It's quite similar to the rectangular distribution but with difference in shape. Again we have here probability we have volume. But now this time the probability of the true calibrated volume being near 10 is higher than being near 10.03 or 9.97 and the shape of the distribution curves looks as a triangle. I deliberately have drawn in such a way that the areas under these two curves are equal. So in both cases the probability of being within this range is 100 % and being outside here is zero but in this case yes as I said the probability of being near ten is higher than away from it. And how do we convert now ? So if we have uncertainty estimate given as plus minus 0.03 then the standard uncertainty estimate of this would be divided by square root of 6 which is equal to roughly 0.012 mL. Now how do we choose which of these two distributions we assume if we do not have information which one is true ? Usually I would recommend such an approach that you choose the way by which the probability of under estimating uncertainty is lower. If we look now then the standard uncertainty estimate of this calibrated volume will be higher if we assume rectangular distribution then if we assume triangular distribution. Therefore as a general rule if you have uncertainty given as plus minus some value and you do not have any information about that uncertainty range I recommend that you assume rectangular distribution. We now look at these two distribution functions only as assumed or postulated distribution functions and you may ask that : "well are there any real cases where rectangular distribution for example really hold ?" And indeed there are. They are not numerous, this distribution does not come up very often but if we speak about rounding of digital reading for example in a digital balance or some other digital meter then the uncertainty due to rounding is strictly uncertainty according to the rectangular distribution and then this dividing by square root of three is a strict operation not an assumption as we have known. How does it work with this rounding uncertainty ? Let us look at a small example. Suppose we have a reading of some instrument let's say pH meter and let's say that reading would be 7.42. So the pH value of some water is 7.42. It's digital reading so we have these two decimals we don't have any other but obviously actually there are more digits that simply are not seen by us because the analog digital converter does not give them to us. So the true reading can be for example 7.418. It can also be 7.424. And it can also be different other values but if we now look carefully it's obvious that this reading certainly will be within the range of… If it were lower than 7.415 we would already see 7.41. If it were higher than this value we would see 7.43. Therefore in this case we know it is between these limits and we have absolutely no information absolutely no preference where it actually is so indeed it is fully reasonable to assume in this case uniform or rectangular distribution. This is also interesting from another point of view. If you remember the introductory lecture of this course I stressed that we almost never can have uncertainty in such a way that some value is strictly within the limits. It turns out now that this particular uncertainty source is strictly within these limits so it cannot be outside. Therefore the probability outside of this rectangle indeed is zero. Though this does not hold always. So if this distribution function is just a postulated distribution function without strict physical background behind it then it is possible that there's also some tiny small probability somewhere but it is assumed to be zero and from practical point of view it does not really matter. |
Examinons désormais quelques autres fonctions de distribution en plus de la distribution normale. Comme je l’ai expliqué, la distribution normale possède une importance unique dans la science de la mesure mais il y en a quelques autres qui sont aussi importants et nous allons maintenant regarder les distributions rectangulaire et triangulaire et nous allons faire cela avec l’exemple de la pipette encore une fois. Nous regardons cette pipette. L’incertitude de calibration de cette pipette est de 0,03 mL et si nous la regardons bien, nous voyons qu’il y a un signe plus/moins devant le 0,03 et il n’y a aucune information donnée ici à propos du statut de cette incertitude sur la fonction de distribution ou sur le facteur de distribution. Regardons comment nous devrions utiliser ce genre d’informations qui viennent d’une façon limitée ce qui veut dire qu’il y a un manque d’informations concrètes sur l’incertitude. La façon habituelle dont les gens gèrent ce genre de situation est que certaines fonctions de distribution sont assumées ou postulées et les distributions triangulaire et rectangulaire sont les plus fréquentes à utiliser dans ce cas. Regardons la distribution rectangulaire. Nous avons ici la probabilité et les valeurs possibles de la mesure et que ce soit le volume calibré de la pipette. Et ici, c’est 10 mL. Et maintenant cette incertitude de ±0,03 sera, ici, comme suit. Et maintenant si c’est une distribution rectangulaire, cela veut dire que la probabilité que le vrai volume calibré soit n’importe où dans cette zone est égale, ce qui veut dire que l’aire sous cette courbe est unitaire. La probabilité que le volume soit à l’extérieur est de zéro et il n’y a aucune distinction entre ces valeurs. Maintenant, si nous voulons utiliser cette incertitude estimée, ce ±0,03 mL dans notre calcul d’incertitude. Nous devons le convertir d’une certaine façon en incertitude type et le convertir depuis la distribution rectangulaire vers une incertitude type est très simple et peut être fait comme suit. C’est simplement divisé par la racine carrée de trois. Donc à chaque fois que nous avons une incertitude exprimée en plus ou moins telle valeur et que nous avons une raison d’assumer la distribution rectangulaire, nous pouvons toujours trouver l’incertitude type estimée en divisant par la racine carrée de 3. Nous allons maintenant regarder la distribution triangulaire. C’est très ressemblant à la distribution rectangulaire mais avec une différence dans la forme. Encore une fois, nous avons ici la probabilité et ici nous avons le volume. Mais cette fois, la probabilité que le vrai volume calibré soit proche de 10 est supérieure à celle d’être proche de 10,03 ou 9,97 et la forme de la courbe de la distribution ressemble à un triangle. J’ai délibérément dessiné de sorte que les aires sous ces deux courbes soient égales. Donc, dans les deux cas, la probabilité d’être dans cet intervalle est de 100% et celle d’être à l’extérieur est de 0 mais dans ce cas, comme je l’ai dit, la probabilité d’être proche de 10 est plus grand que d’en être loin. Et comment nous convertissons maintenant ? Donc si nous avons une incertitude estimée donnée sous forme de plus ou moins 0,03 alors l’incertitude type estimée de cela serait divisée par la racine carrée de 6, pour un résultat égal grossièrement à 0,012 mL. Maintenant, comme nous choisissons laquelle de ces deux distributions nous utilisons si nous n’avons pas d’informations sur laquelle est vraie ? Habituellement, je recommanderai une approche telle que vous choisissez la façon pour laquelle la probabilité de sous-estimer l’incertitude est la plus basse. Si nous regardons maintenant, l’estimation de l’incertitude type de ce volume calibré sera plus élevée si nous utilisons la distribution rectangulaire plutôt que si nous utilisons la distribution triangulaire. Par conséquent, comme une règle générale si vous avez une incertitude donnée sous forme de plus ou moins une valeur et que vous n’avez pas d‘informations sur l’intervalle d’incertitude, je vous recommande d’utiliser la distribution rectangulaire. Nous allons maintenant regarder ces deux fonctions de distribution seulement comme des fonctions de distribution assumées ou postulées et vous pouvez vous demander : « est-ce qu’il y a des vrais cas où la distribution rectangulaire peut réellement fonctionner ? ». Et en effet, il y en a. Ils ne sont pas nombreux, cette distribution n’est pas utilisée très souvent mais si nous parlons d’arrondis de la lecture numérique, par exemple sur une balance numérique ou sur d’autres compteurs numériques alors l’incertitude liée à l’arrondissement est strictement une incertitude fonction de la distribution rectangulaire et par conséquent, cette division par la racine carrée de trois est une opération rigoureuse et pas une supposition comme nous le savions. Comment cela fonctionne avec une incertitude d’arrondis ? Nous allons regarder un petit exemple. Supposons que nous avons une lecture d’un quelconque instrument, disons un pH-mètre et disons que la valeur lue est 7,42. Donc la valeur de pH d’une certaine eau est 7,42. C’est une lecture numérique donc nous avons ces deux décimales et nous n’en avons pas d’autres mais évidemment en réalité il y a plus de chiffres mais ils ne sont simplement pas vus par nous parce que le convertisseur numérique analogique ne nous les donne pas. Par conséquent, la vraie valeur peut être par exemple 7,418. Elle peut être aussi 7,424. Et elle peut être aussi d’autres valeurs mais si maintenant nous regardons attentivement, il est évident que cette lecture sera dans l’intervalle entre… Si c’était plus petit que 7,415 nous verrions 7,41. Si c’était plus grand que cette valeur, nous verrions 7,43. Par conséquent, dans ce cas nous savons qu’elle est entre ces deux limites et nous avons absolument aucune information, absolument aucune préférence de où elle est vraiment, donc en effet il est totalement raisonnable d’admettre, dans ce cas une distribution uniforme ou rectangulaire. Ceci est également intéressant depuis un autre point de vue. Si vous vous souvenez la leçon d’introduction de ce cours, j’ai souligné que nous ne pouvons presque jamais avoir une incertitude de telle sorte qu’une certaine valeur est strictement entre ces limites. Il s’avère maintenant que cette source d’incertitude particulière est strictement entre ces limites donc elle ne peut pas être à l’extérieur. Par conséquent, la probabilité d’être à l’extérieur de ce rectangle est en effet de zéro. Bien que cela ne tienne pas toujours. Donc si cette fonction de distribution est juste une fonction de distribution postulée sans un arrière-plan physique rigoureux derrière alors il est possible qu’il y ait aussi une toute petite probabilité quelque part mais il est admis qu’elle est de zéro et depuis ce point de vue pratique ce n’est pas vraiment important. |
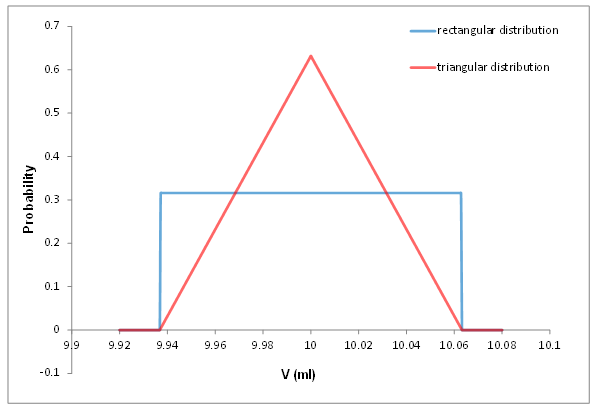
Distributions rectangulaires et triangulaires.
Les deux distributions correspondent à la situation (10.000 ± 0,063) ml.
Sur la base d'une distribution rectangulaire, l'incertitude type peut être calculée par l'incertitude divisée par racine de 3
Sur la base d'une distribution triangulaire, l'incertitude type peut être calculée par l'incertitude divisée par racine de 6.
L’estimation de l’incertitude de mesure conduit souvent à choisir entre deux options, l’une pouvant conduire à une incertitude quelque peu surestimée et l’autre à une incertitude légèrement sous-estimée. Dans une telle situation, il est généralement raisonnable de surestimer plutôt que de sous-estimer l’incertitude.
Autoévaluation sur cette partie du cours : Test 3.5
4.6. La distribution de Student
https://sisu.ut.ee/measurement/35-other-distribution-functions-student-distribution
Résumé : Comme les valeurs individuelles, la valeur moyenne calculée à partir de celles-ci est également une quantité aléatoire. Si les valeurs individuelles sont réparties selon la loi de distribution normale, la valeur moyenne calculée à partir de celles-ci est répartie selon la distribution de Student (également appelée distribution t). Les propriétés de la distribution t comparées à la distribution normale sont expliquées. Il est important de noter que la forme de la courbe de distribution t dépend du nombre de degrés de liberté. Si le nombre de valeurs individuelles approche l'infini, la forme de la courbe de distribution t se rapproche de la courbe de distribution normale.
La distribution de Student
http://www.uttv.ee/naita?id=17708
| Let us now examine one more distribution function : the T or so-called Student distribution. Student distribution is somewhat similar to the normal distribution in that it is also characterized by a mean value and the standard deviation. But in addition to these two, it is also characterized by the number of degrees of freedom, which makes it different from the normal distribution function. When do we encounter Student distribution ? The Student distribution is encountered first of all in all those cases where we use mean values, so whenever we carry out repeated measurements of the same quantity and we use the mean value of those repetitions. The mean value also is a random quantity and also obeys a distribution function. and the mean value of normally distributed values is distributed according to the Student distribution. And let us go for a moment back to the normal distribution. We have examined that quite carefully and we have discovered that these probabilities here are quite important because they set for us the coverage probabilities of the different uncertainties. So for example, standard uncertainty has the coverage probability of roughly 68% and k2 expanded uncertainty has the coverage probability of roughly 95% if the result is distributed according to the normal distribution. When I explained that, I already mentioned that there will be cases where this 95 percent will not hold if we use k2 expanded uncertainty. In actually one of the main reasons, that it does not always hold, is the Student distribution. And let us see how it is done and let us see what the number of the degrees of freedom means. So the number of degrees of freedom is equal to the number of parallel measurements minus one. So if the mean value is obtained from 101 measurements, then the number of degrees of freedom is 100. If from 11 measurements it's 10, if from 6 measurements it's 5 and if it is obtained from 3 measurements only, then the number of degrees of freedom is 2. Now the Student distribution has an interesting property that whenever the number of degrees of freedom approaches infinity, the Student distribution approaches the normal distribution. And for all practical cases, we can see that, let's say from 50 to 30 measurements, give us distribution function which is almost indistinguishable from the normal distribution. Therefore, the normal distribution curve here, which corresponds to 100 degrees of freedom, can be regarded as the normal distribution. Therefore, the student distribution curve here, which is obtained from 101 measurements, can be regarded as a normal distribution. But now, as the number of degrees of freedom decreases, which is the consequence of lower number of repeated measurements, lower number of parallel measurements, the shape of the distribution function changes and the shape changes interestingly. So you see here, in the center, the intensity decreases so that the probability of the true mean value being here decreases somewhat, but at the same time, the tails, the tails become higher and higher more powerful. Therefore, we can say that the probability to some extent is flowing away from the middle into the tails and this is what causes, that if our result is distributed according to the normal distribution, then k2 uncertainty can be uncertainty with much less probability than 95%. And we can look at it, this way, if it is the normal distribution, then the area of this part here plus the area of this part here is roughly 4.5%, which is the probability that remains outside of this 95.5%. But we see that, as the number of degrees of freedom decreases, the tail gets more and more and more intense, meaning the area here gets larger and larger. Therefore, whenever we have lower number of degrees of freedom, the probability that we can achieve by the k2 expanded uncertainty becomes lower than 95%. It can be 93, 92 even 85% if the number of degrees of freedom is small. Now, interestingly, as we will also see and comment on in some of the examples, usually our measurements results are distributed neither normally nor according to the Student distribution. Their distribution usually is the convolution of different distributions. And as I have been explaining already, whenever our measurement result is influenced by many different input quantities, then if each of those input quantities has their own distribution function, then if the number of those quantities or influencing factors approaches infinity, then the distribution function approaches the normal distribution, so that if the measurement result is influenced by many many factors then usually we can assume that it is distributed normally. In reality however, very often it happens that there is one, two or three strong influencing factors and the remaining ones are not so strong. And in such a case, the strong factors determine the shape of the distribution function of the output quantity. And if one of those strong influencing factors has T distribution with low number of degrees of freedom, then our result will be distributed according to a convolution of normal and T distribution and in such a case, the k2 uncertainty corresponds to coverage probability which is less than 95%. In practice, such situation most often occurs if there is an important and influential input quantity which is obtained as mean value of, let's say, three, four or five measurements. So if three, four or five measurements are averaged into a mean value, and this mean value has an important influence on the measurement uncertainty, then we can be quite sure that the result is distributed not according to purely normal distribution but by a mixed distribution and convolution of the normal and Student distribution. |
Examinons maintenant une autre fonction de distribution : la T ou la dite distribution de Student. La distribution de Student est assez similaire à la distribution normale dans le sens où elle est aussi caractérisée par une valeur moyenne et un écart-type. Mais en plus de ces deux valeurs, elle est aussi caractérisée par le nombre de degrés de liberté, ce qui la rend différente de la fonction de distribution normale. Quand rencontrons-nous la distribution de Student ? La distribution de Student est rencontrée tout d'abord dans tous les cas où nous utilisons des valeurs moyennes, donc lorsque nous effectuons des mesures répétées de la même quantité et que nous utilisons la valeur moyenne de ces répétitions. La valeur moyenne est également une quantité aléatoire et obéit aussi à une fonction de distribution. La valeur moyenne des valeurs distribuées normalement est distribuée selon la distribution de Student. Revenons un instant à la distribution normale. Nous avons examiné cela très attentivement et nous avons découvert que ces probabilités ici sont plutôt importantes car elles nous posent les probabilités de couverture des différentes incertitudes. Ainsi, par exemple, l'incertitude type a une probabilité de couverture d'environ 68% et l'incertitude élargie k2 a une probabilité de couverture d'environ 95% si le résultat est distribué selon la distribution normale. Lorsque j'ai expliqué cela, j'ai déjà mentionné qu'il y aurait des cas où ces 95% ne tiendront pas si nous utilisons l'incertitude élargie k2. En fait, l'une des raisons principales, qui ne tient pas toujours, est la distribution de Student. Voyons comment cela se fait et que signifie le nombre de degrés de liberté. Le nombre de degrés de liberté est égal au nombre de mesures parallèles moins un. Donc si la valeur moyenne est obtenue à partir de 101 mesures, alors le nombre de degrés de liberté est 100. À partir de 11 mesures c'est 10, à partir de 6 mesures c'est 5 et si elle est obtenue à partir de 3 mesures seulement, alors le nombre de degrés de liberté est 2. Maintenant, la distribution de Student possède une propriété intéressante qui est qu'à chaque fois que le nombre de degrés de liberté approche l'infini, la distribution de Student approche la distribution normale. Pour tous les cas pratiques, nous pouvons voir que, disons pour 50 à 30 mesures, cela nous donne une fonction de distribution qui est presque non distinguable de la distribution normale. Par conséquent, la courbe de distribution normale ici, qui correspond à 100 degrés de liberté, peut être considérée comme une distribution normale. Par conséquent, la courbe de distribution de Student ici, qui est obtenue à partir de 101 mesures, peut être considérée comme une distribution normale. Mais maintenant, comme le nombre de degrés de liberté décroit, ce qui est la conséquence d'un nombre inférieur de mesures répétées, d'un nombre inférieur de mesures parallèles, la forme de la fonction de distribution change et elle change de façon intéressante. Vous pouvez donc voir ici, au centre, que l'intensité diminue de sorte que la probabilité de la valeur moyenne vraie ici décroit quelque peu, mais dans un même temps, les queues deviennent de plus en plus puissantes. C'est pourquoi, nous pouvons dire que la probabilité, dans une certaine mesure, s'éloigne depuis le centre vers les queues et cela implique, que si notre résultat est distribué selon la distribution normale, alors l'incertitude k2 peut être une incertitude avec une probabilité bien inférieure à 95%. Nous pouvons l'examiner de cette manière : si c'est une distribution normale, alors l'aire de cette partie ici plus l'aire de cette partie là est d'environ 4,5%, ce qui est la probabilité qui reste en dehors de 95,5%. Mais nous voyons que, comme le nombre de degrés de liberté décroit, la queue devient de plus en plus intense, ce qui signifie que l'aire devient de plus en plus large. De fait, chaque fois que nous avons un nombre de degré de liberté plus faible, la probabilité que nous pouvons atteindre avec l'incertitude élargie k2 devient inférieure à 95%. Ça peut être 93, 92, même 85% si le nombre de degré de liberté est petit. Maintenant, il est intéressant de noter que, comme nous le verrons et commenterons dans certains exemples, nos résultats de mesures ne sont généralement pas distribués normalement ni selon la distribution de Student. Leur distribution est généralement la convolution de différentes distributions. Comme je l'ai déjà expliqué, lorsque notre résultat de mesure est influencé par de nombreuses quantités d'entrée différentes, si chacune de ces quantités d'entrée a sa propre fonction de distribution, si le nombre de ces quantités ou de facteurs d'influence approche l'infini, alors la fonction de distribution approche la distribution normale, de sorte que si le résultat de mesure est influencé par de nombreux facteurs, alors nous pouvons généralement supposer qu'il est distribué normalement. En réalité cependant, il arrive très souvent qu'il y ait un, deux ou trois facteurs d'influence forts et que les autres ne le soient pas autant. Dans ce cas, les facteurs forts déterminent la forme de la fonction de distribution de la quantité de sortie. Et si un de ces facteurs d'influence forts a une distribution T avec un faible nombre de degrés de liberté, alors notre résultat sera distribué selon une convolution de distribution normale et T et dans un tel cas, l'incertitude k2 correspond à une probabilité de couverture qui est inférieure à 95%. En pratique, une telle situation se produit le plus souvent s'il y a une quantité importante et influente d'entrée qui est obtenue en tant que valeur moyenne de, disons trois, quatre ou cinq mesures. Donc si trois, quatre ou cinq mesures sont moyennées en une valeur moyenne, et que cette valeur moyenne a une influence importante sur l'incertitude de mesure, alors nous pouvons être assez sûrs que le résultat est distribué non pas selon une distribution purement normale mais par une distribution et convolution mixte d'une distribution normale et une distribution de Student. |
Si une mesure est répétée et que la moyenne est calculée à partir des résultats des mesures individuelles, alors, tout comme les résultats individuels, leur moyenne sera également une quantité aléatoire. Si les résultats individuels sont normalement distribués, leur moyenne est répartie selon la distribution de Student (également appelée distribution t). La distribution de Student est présentée dans le schéma 3.5.
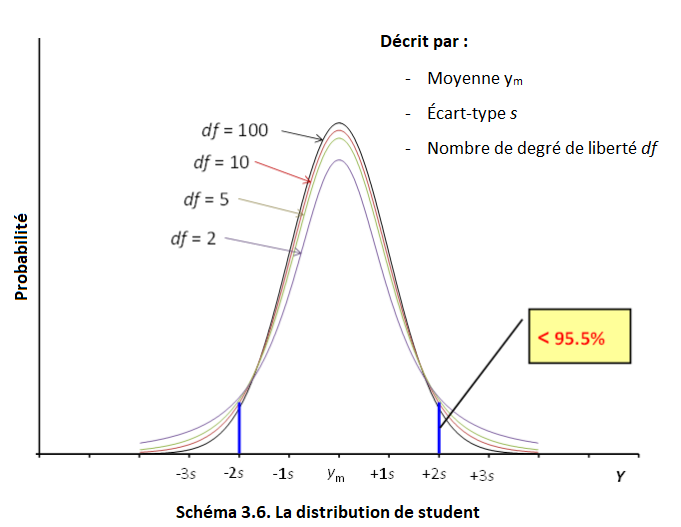
De manière similaire à la distribution normale, la distribution de Student a également une valeur moyenne ym et un écart type s. ym est la valeur moyenne elle-même [1] et l'écart-type est l'écart-type de la moyenne, calculé comme expliqué à la section 3.4. Mais, à la différence de la distribution normale, il existe une troisième caractéristique : le nombre de degrés de liberté df. Ce nombre est égal au nombre de mesures répétées moins un. Ainsi, les quatre graphiques de distribution de Student du schéma 3.5 correspondent respectivement à 101, 11, 6 et 3 mesures répétées.
Si df approche l'infini, la distribution t se rapproche de la distribution normale. En réalité, 30 à 50 degrés de liberté suffisent pour gérer la distribution t comme distribution normale. Ainsi, la courbe avec df = 100 dans le schéma 3.5 peut être considérée comme la courbe de distribution normale.
Plus le nombre de degrés de liberté est faible, plus les «queues» de la courbe de distribution de Student sont «lourdes» et plus la distribution est différente de la distribution normale. Cela signifie que plus de probabilité réside dans les queues de la courbe de distribution et moins dans la partie médiane. Il est important de noter que les probabilités représentées dans le schéma 3.2 pour les plages de ± 1, ± 2 et ± 3 s autour de la moyenne ne sont plus adaptées, mais sont toutes plus basses.
Ainsi, si un résultat de mesure est distribué selon la distribution t et si une incertitude élargie avec une probabilité de couverture prédéfinie est souhaitée, alors, au lieu des facteurs de couverture habituels 2 et 3, il convient d'utiliser les coefficients de Student respectifs [2]. Le résultat de la mesure peut être distribué en fonction de la distribution de Student s'il existe une source fortement prédominante [3] d'incertitude de type A qui a été évaluée en tant que valeur moyenne à partir d'un nombre limité de mesures répétées. Cependant, il est plus courant de constater qu’une source d’incertitude de type A, influente mais pas très dominante, existe. Dans un tel cas, la distribution du résultat de la mesure est une convolution [4] de la distribution normale et de la distribution t. La procédure à suivre dans ce cas est expliquée à la section 9.8.
Autoévaluation sur cette partie du cours : Test 3.6
https://sisu.ut.ee/measurement/measurement-1-4
***
[1] Il peut sembler étrange à première vue que, bien que la valeur moyenne ym soit la seule valeur moyenne dont nous disposions, nous la prenions immédiatement comme la valeur moyenne de la distribution des valeurs moyennes. Cependant, si nous avions plus de valeurs moyennes, nous les regrouperions dans une seule valeur moyenne (avec un df beaucoup plus élevé) Et utiliserions cette valeur.
[2] Les coefficients de Student (c.-à-d. Les valeurs de la distribution t) pour un ensemble donné de probabilité de couverture et le nombre de degrés de liberté peuvent être facilement obtenus à partir de tableaux spéciaux de manuels statistiques (utilisez des valeurs bilatérales!), À l'aide d'un logiciel de calcul ou de traitement de données. , tels que MS Excel ou Openoffice Calc ou sur Internet, par exemple de l'adresse https://en.wikipedia.org/wiki/Student_distribution
[3] Les contributions de différentes sources d'incertitude peuvent être exprimées numériquement. Ceci est expliqué dans la section 9.9 et les calculs respectifs sont présentés dans 9.7. Dans ce contexte, l'expression "fortement dominant" signifie que la contribution (indice d'incertitude) de la quantité d'entrée respective est supérieure à 75%.
[4] La convolution de deux fonctions de distribution en statistique mathématique désigne une combinaison de fonction de distribution, qui a une forme intermédiaire entre les deux fonctions de distribution convolutives.
5. Première quantification de l'incertitude
https://sisu.ut.ee/measurement/4-principles-measurement-uncertainty-estimation
Résumé : Dans cette section, les concepts de base et les outils des sections précédentes sont mis en pratique à travers l'exemple d'une opération de chimie analytique simple : le pipetage. Les sources d'incertitude identifiées dans la section 2 sont maintenant quantifiées (4.1), les estimations individuelles d'incertitude obtenues sont (le cas échéant) converties en incertitudes types, puis combinées en incertitude-type composée (4.2). Les composantes de l'incertitude constituant l'incertitude-type composée sont comparées et certaines conclusions sont tirées (4.3). L'incertitude-type composée est convertie en incertitude élargie (4.4) et le résultat est présenté (4.5). Cette approche est ensuite mise en pratique sur un exemple de calcul (calibration de la pipette, 4.6).
5.1. Quantification des sources d'incertitude
https://sisu.ut.ee/measurement/41-quantifying-uncertainty-components
Résumé : Le même pipetage que dans le chapitre 2 est maintenant examiné du point de vue de la quantification des sources d’incertitude. Toutes les sources d'incertitude importantes sont exprimées de manière quantitative en tant que composantes d'incertitude (estimations d'incertitude décrivant quantitativement la source d'incertitude respective). Les composantes de l'incertitude sont quantifiées. On donne un exemple pour convertir une estimation d'incertitude avec une distribution (supposée) rectangulaire, c'est-à-dire uniforme, en une estimation d'incertitude type.
Introduction à la quantification de l'incertitude de mesure
http://www.uttv.ee/naita?id=17555
|
Let us now try to make our first uncertainty quantification. We will look at pipetting again, and we will look at the volume that we pipetted in one of the previous videos. We will first look at what are the uncertainty sources that affect this pipetting. As I mentioned already in that video, they are in broad terms the following. If we look at the volume then, it is affected by the uncertainty of repeatability of pipetting. It is also affected by the uncertainty due to the calibration of the pipette, meaning the uncertainty with which the mark is exactly on 10 mL position. But also is affected by the temperature effect. Remember, water density depends on temperature. Therefore at different temperatures, actually different amounts of water meaning different numbers of water molecules are dispensed. There are some other sources of uncertainty that can affect pipetting but which are not really important in our case. But for the sake of completeness I also will bring this here. We'll use dotted line to show that we don't really need to take them into account now. These are first of all, if the liquid is very different from the liquid with which the pipette was calibrated. Secondly, if the pipette is not clean, the walls will develop droplets on them. So, now we have our uncertainty sources. That's usually how the uncertainty evaluation goes. Those uncertainty sources are quantitatively expressed by uncertainty components. So, to each and every of them, we have an uncertainty component. So, the standard uncertainty of this volume, due to repeatability. This is how this uncertainty component reads. Standard uncertainty due to calibration and standard uncertainty due to the temperature effect. You notice that I'm using the term "standard uncertainty" throughout now. This is because, for uncertainty evaluation, we always need to transfer, to convert, all our uncertainty components to the standard uncertainty. Let us look now how do we quantify this uncertainty components. From the data that we have, from our pipetting operation, we have information about them. First of all, the repeatability part, as we saw in one of the previous lectures, can be simply calculate by calculated by the standard deviation of the individual pipetting operations. In this particular case, this repeatability standard deviation turned out to be 0.006 mL. Now, the temperature effect is somewhat more complex to calculate. We will look at that separately, but let us here only say that, with our data, the temperature effect amount to 0.005 mL. Finally, with the calibration part, or with the calibration component, as we saw on the pipette: the pipetted volume deferred from, or can differ, from the nominal volume by as much as plus minus 0.03 mL. This now is not a standard uncertainty estimate. There is no information on that pipette. What actually is the level or what actually is the content of this uncertainty source? As we saw in the lecture where I touched rectangular distribution, it is very reasonable to assume that this volume has also rectangular distribution, meaning the volume can be anywhere within 10 plus minus 0.03 mL. In order to convert this uncertainty estimate into a standard uncertainty estimate, we divided by the square root of 3. So 0.03 mL divided by square root of 3 which is 0.017 mL. |
Essayons maintenant de faire notre première quantification de l'incertitude. Nous examinerons à nouveau le pipetage et nous examinerons le volume que nous avons pipeté dans l'une des vidéos précédentes. Nous allons d'abord voir quelles sont les sources d'incertitude qui affectent ce pipetage. Comme je l'ai déjà mentionné dans cette vidéo, ils sont en termes généraux les suivants. Si nous regardons alors le volume, il est affecté par l'incertitude de répétabilité du pipetage. Il est également affecté par l'incertitude due à l'étalonnage de la pipette, c'est-à-dire l'incertitude avec laquelle la marque est exactement à une position de 10 mL. Mais il est également affecté par l'effet de la température. N'oubliez pas que la densité de l'eau dépend de la température. C'est pourquoi à différentes températures, différents volumes d'eau et donc différents nombres de molécules d'eau sont réellement distribués. Il existe d'autres sources d'incertitude qui peuvent affecter le pipetage mais qui ne sont pas vraiment importantes dans notre cas. Mais par souci d'exhaustivité, je vais également en parler ici. Nous allons utiliser une ligne pointillée pour montrer que nous n'avons pas vraiment besoin de les prendre en compte maintenant. Tout d'abord, si le liquide est très différent du liquide avec lequel la pipette a été calibrée. Deuxièmement, si la pipette n'est pas propre, les parois vont retenir des gouttelettes. Nous avons donc maintenant nos sources d'incertitude. C'est généralement de cette façon que ce fait l'évaluation de l'incertitude. Ces sources d'incertitude sont exprimées quantitativement par des composantes d'incertitude. Donc, pour chacune d'entre elles, nous avons une composante d'incertitude. Donc, l'incertitude type de ce volume, due à la répétabilité. C'est ainsi que se lit cette composante d'incertitude. L'incertitude type due à l'étalonnage et l'incertitude type due à l'effet de la température. Vous remarquez que j'utilise le terme "incertitude type" depuis le début. En effet, afin d'évaluer l'incertitude, nous devons toujours transférer, convertir, toutes nos composantes d'incertitude en une incertitude type. Regardons maintenant comment quantifier ces composantes d'incertitude. À partir des données que nous avons, de notre opération de pipetage, nous avons des informations à leur sujet. Tout d'abord, concernant la partie répétabilité, comme nous l'avons vu dans l'un des précédents chapitres, elle peut être calculée simplement par calcul de l'écart type des pipetages individuels. Dans ce cas particulier, cet écart-type de répétabilité s'est avéré être de 0,006 mL. Maintenant, l'effet de la température est un peu plus complexe à quantifier. Nous allons examiner cela séparément, mais disons seulement ici qu'avec nos données, l'effet de la température s'élève à 0,005 mL. Enfin, pour la partie étalonnage, ou la composante étalonnage, comme nous l'avons vu sur la pipette : le volume prélevé par la pipette diffère ou peut différer du volume théorique de plus ou moins 0,03 mL. Il ne s'agit plus maintenant d'une estimation de l'incertitude type. Il n'y a aucune information sur cette pipette. Quel est le réel niveau ou quel est réellement le contenu de cette source d'incertitude ? Comme nous l'avons vu dans le cours où j'ai abordé la loi uniforme continue, il est très raisonnable de supposer que ce volume suit également la loi uniforme continue, ce qui signifie que le volume réel se situe dans un encadrement de plus ou moins 10 mL autour de 0,03. Afin de convertir cette estimation d'incertitude en une estimation d'incertitude type, on divise par la racine carrée de 3. Donc 0,03 mL divisé par la racine carrée de 3 donne alors 0,017 mL. |
Les principales sources d’incertitude sont les mêmes que celles expliquées dans le chapitre 2 et ici les composantes de l’incertitude leur correspondant sont quantifiées.
- Incertitude due à la répétabilité non idéale, ce qui dans le cas du pipetage signifie que, même avec un minimum de soin pour remplir et vider la pipette, nous obtiendrons néanmoins à chaque fois un volume de pipette légèrement différent. La répétabilité est un effet aléatoire typique. L'incertitude type due à la répétabilité u (V, REP) peut être calculée en tant qu'écart-type [1] d'exactitude du volume délivré par la pipette. [2] Selon les données de la pipette utilisée.
image perdue (4.1)
- Incertitude due à l'étalonnage de l'équipement volumétrique (souvent appelée incertitude d'étalonnage ou incertitude du volume nominal). C'est l'incertitude dans la position des marques sur les articles volumétriques. Dans le cas d'une pipette donnée, il s'agit d'un effet systématique typique. L'incertitude d'étalonnage de la pipette utilisée dans cet exemple est spécifiée par le fabricant à ± 0,03 ml. [3] Il n'existe aucune information sur la distribution ou la couverture de cette estimation d'incertitude. Ceci est très courant si les estimations d'incertitude sont obtenues à partir de la documentation de l'instrument. Dans ce cas, le plus sûr est de supposer que l'estimation de l'incertitude correspond à une distribution rectangulaire. Afin de pouvoir calculer l'incertitude, nous devons convertir cette incertitude en incertitude type. Pour se faire, comme expliqué dans la section 3.5, nous devons le diviser par la racine carrée de 3.
image perdue (4.2)
- Incertitude due à l'effet de la température (souvent appelée incertitude de la température). Tous les appareils volumétriques sont généralement calibrés à 20 ° C et les volumes se réfèrent généralement à des volumes à 20°C [4]. Le changement de température affecte tout d'abord la densité du liquide (l'effet de dilatation / contraction du verre est considérablement réduit). Si le pipetage est effectué à une température supérieure à 20°C, la quantité de liquide pipetée (en masse ou en nombre de molécules) est inférieure à celle correspondant au volume à 20°C. Par conséquent, le volume de cette quantité de liquide à 20°C est également plus petit que si le pipetage était effectué à 20°C. Si la température du laboratoire et, ce qui est important, la température du liquide pipetté, est constante pendant les mesures répétées, alors l'effet de la température est un effet systématique. La vidéo suivante explique comment calculer l’incertitude type du volume de liquide causée par la température u (V, TEMP).
Quantifier l'incertitude due à l'effet de la température dans les mesures volumétriques
http://www.uttv.ee/naita?id= 17825
|
Let us look now how to quantify the uncertainty due to the temperature effect in volumetric measurement. The main equation for such uncertainty calculation reads as follows: standard uncertainty of a certain volume due to temperature is equal to: first of all, the volume itself, V. Then this is multiplied by the maximum possible deviation of the temperature from 20°C, which we denote here by delta t. And this term is multiplied by the thermal expansion coefficient of water, which we denote by gamma. And now if this delta t or the maximum deviation can be estimated as a rectangularly distributed meaning, the temperature of the water can be anywhere within this range. Then we also need to divide this all expression by scare root of three in order to convert this rectangular distribution into a normal distribution based standard uncertainty. So, we write here: square root of three. And now we can put in the data that we had in our specific example. The volume of our pipette was 10 mL. The maximum possible difference of temperature from 20°C centigrade, we estimate as plus minus 4°C. And the value of thermal expansion coefficient of water is 0.00021 to reciprocal degrees. And if we calculate the value of uncertainty due to the temperature effect then what we find is 0.0049 mL. |
Nous allons maintenant voir comment quantifier l’incertitude due à l’effet de la température dans le cas des mesures volumétriques. La principale équation pour ce calcul d’incertitude est la suivante : l’incertitude type, d'un certain volume, due à la température est égale à : tout d’abord, le volume en lui-même, V. Puis, cela est multiplié par l’écart maximum de la température par rapport à 20°C, que l’on note delta t. Et cela, est multiplié par le coefficient de dilatation thermique de l’eau, que l’on note gamma. Maintenant, si ce delta t ou l’écart maximum peut être estimé suivant une loi uniforme continue, la température de l’eau peut se situer n’importe où dans cette plage. Puis, il est également nécessaire de diviser la totalité de cette expression par la racine carrée de trois afin de convertir cette loi uniforme continue en une loi de répartition normale pour obtenir l’incertitude type. Donc, on écrit ici la racine carrée de trois. Enfin, nous pouvons insérer les données que nous avons à partir de notre exemple. Le volume de notre pipette est de 10 mL. La différence maximale de température par rapport à 20°C, est estimée à plus ou moins 4°C. Et, la valeur du coefficient de dilatation thermique de l’eau est de 0,00021 aux chiffres significatifs près. Finalement, si nous calculons la valeur de l’incertitude due à l’effet de la température on obtient 0,0049 mL. |
u (V, TEMP) dépend du volume V de liquide fourni [5], de la différence de température maximale possible à partir de 20°C (Δt) et du coefficient de dilatation thermique de l'eau γ. Il est calculé comme suit :
image perdue (4.3)
La division par la racine carrée de 3 sert à transformer l'estimation de l'incertitude en incertitude type(en supposant une distribution rectangulaire de Δt). Il est important de noter que le V dans l'équation 4.3 fait toujours référence au volume réellement mesuré, et non à la capacité totale du dispositif volumétrique. Par exemple, si on a mesuré 21,2 ml de solution avec une burette de 50 ml, le volume à utiliser est de 21,2 ml et non de 50 ml.
Dans le cas d'estimations d'incertitude plus complexes, certaines des sources peuvent également être présentées avec une incertitude élargie. Par exemple, la teneur en analyte dans le matériau de référence: (0,2314 ± 0,0010) mg/kg, k = 2. Dans ce cas, cette incertitude doit également être convertie en incertitude type pour des calculs ultérieurs. Pour cela, l'incertitude élargie est divisée par le facteur d'élargissement présenté. Ainsi, dans l'exemple donné, l'incertitude type composée vaut 0,0005 mg/kg.
Autoévaluation sur cette partie du cours : Test 4.1
https://sisu.ut.ee/measurement/node/1522 (lien mort)
***
[1] Etant donné que le pipetage pour délivrer un certain volume de liquide n’est effectué qu’une seule fois et ne peut faire l’objet d’une moyenne (c’est-à-dire qu’il n’est pas possible de pipeter plusieurs fois puis de "moyenner" les volumes), l’estimation appropriée de l’incertitude de répétabilité est l’écart type associé à une mesure, pas l'écart type de la moyenne.
[2] Les volumes exacts peuvent être mesurés en pesant l’eau fournie par la pipette et en la convertissant en volume en utilisant des données de densité exactes.
[3] Cette incertitude peut être considérablement réduite si la pipette est recalibrée en laboratoire en pesant l'eau fournie.
[4] Si la verrerie volumétrique est étalonnée dans le même laboratoire, une température différente peut être utilisée.
[5] Dans le cas d'une pipette volumétrique, le volume nominal est identique au volume délivré, mais dans le cas d'une burette, il ne l'est généralement pas. Ainsi, si vous administrez 12,63 ml de solution d'une burette de 25 ml, le volume à utiliser pour le calcul de l'effet de la température est de 12,63 ml et non de 25 ml.
5.2. Calcul de l'incertitude-type composée
https://sisu.ut.ee/measurement/42-combining-uncertainty-components-combined-standard-uncertainty
Les composantes de l'incertitude qui ont été quantifiées lors du chapitre précédent sont maintenant combinées dans l'incertitude type composée (uc) - incertitude type qui prend en compte les contributions de toutes les sources d'incertitude importantes en combinant les composantes respectives de l'incertitude. Le concept de mesure indirecte - par lequel la valeur de la quantité de sortie (résultat de mesure) est trouvée par une fonction (modèle) à partir de plusieurs quantités d'entrée - est introduit et expliqué. La majorité des mesures chimiques sont des mesures indirectes. Le cas général de la combinaison des composantes de l’incertitude en une incertitude type composée ainsi que plusieurs cas spécifiques sont présentés et expliqués.
La première vidéo explique de manière simple comment les composantes de l'incertitude sont combinées dans l'exemple particulier du pipetage. La deuxième vidéo présente l’aperçu général de la combinaison des composantes de l’incertitude.
Combinaison des composantes de l'incertitude dans l'incertitude type composée dans le cas du pipetage
http://www.uttv.ee/naita?id=17556
|
We have seen now how to quantify the individual uncertainty components of the pipetted volume and now we will see how do we combine those individual uncertainty components into the overall uncertainty of volume that would then take into account the contributions of all those uncertainty sources that are characterized by those uncertainty components. And this overall uncertainty of volume we denote by uc(V). U still stands for the standard uncertainty as it was previously but C is a new thing and it means "combined" so this uc(V) means combined standard uncertainty of the volume. This means that it combines together all the different uncertainty sources. It can be calculated in different ways as we see later on but today we can use the simplest possible approach which works as follows. So, we take all these individual uncertainty components. We take all of them to the square and we sum up all those squares and take square root. This is sometimes called squared sum or squared summing. So that this is the way how we calculate the combined standard uncertainty of the volume. And now we will put concrete values here and let's see what we will get. Here they are and the combined standard uncertainty will be 0.019 mL. |
Nous avons vu comment quantifier les incertitudes individuelles des volumes pipetés et nous allons voir à présent comment nous pouvons combiner ces incertitudes individuelles en une incertitude totale de volume qui prendrait en compte les contributions de toutes les sources d'incertitudes caractérisées par ces incertitudes individuelles. Et cette incertitude totale de volume s'écrit Uc(V). U correspond toujours à l'incertitude type comme on l'a vu précédemment, mais C'est une nouveauté et signifie "composée" donc Uc(V) veut dire incertitude type composée de volume. Cela signifie qu'elle combine l'ensemble des différentes sources d'incertitudes différentes. Elle peut être calculée de différentes manières comme nous pourrons le voir plus tard, mais aujourd'hui nous pouvons utiliser l'approche la plus simple possible qui fonctionne de la manière suivante. Donc, nous prenons toutes ces composantes d'incertitudes individuelles. Nous les mettons toutes au carré et nous les sommons toutes puis nous prenons la racine. Cela est parfois appelé la somme des carrés. Donc c'est la manière dont on calcule l'incertitude type composée de volume. Et maintenant nous allons utiliser des valeurs concrètes et voir ce que nous allons obtenir. Et voilà, l'incertitude-type composée sera 0,019 mL. |
Dans tous les cas où l'incertitude type composée est calculée à partir de composantes d'incertitude, toutes les composantes d'incertitude doivent être converties en incertitudes-type.
Dans l'exemple de pipetage, l'incertitude-type composée est calculée à partir des composantes d'incertitude décrites dans la section précédente, comme suit:
image perdue (4.4)
C'est la manière typique de calculer l'incertitude-type composée si toutes les composantes de l'incertitude se réfèrent à la même quantité et sont exprimées dans les mêmes unités. Il est souvent utilisé dans le cas de mesures directes - mesures pour lesquelles l'instrument de mesure (pipette dans ce cas) donne immédiatement la valeur du résultat, sans autres calculs.
Combinaison des composantes de l'incertitude dans l'incertitude-type composée : cas simple et cas général
http://www.uttv.ee/naita?id=17826
|
Let us now look at the key concept of measurement uncertainty estimation : the combined standard uncertainty. If we speak about measurements, the measurement results are almost always influenced by different factors or they can be calculated from different parameters or quantity. And so, as it is called in measurement science, our measurement result is an output quantity which is influenced by, or calculated from, a number of input quantities. And now the combined standard uncertainty is the uncertainty of the output quantity which takes into account the uncertainties of all the input quantities. And combined standard uncertainty as we already saw in the previous lecture is denoted by uc whereby C means combined. And most of the measurements in chemistry are indirect measurements meaning that we do not see from our measurement instrument display immediately the result, the answer, but rather we measure different quantities and different parameters and calculate from their values our measurement result value. And all such measurements were from values of input quantities and the output quantities calculated are called indirect measurements. And the good example is titration. When we do titration we need to know titrant concentration, we need to know the volume of our sample solution that we took for titration and we also need to know the volume of titrant that was consumed for this titration. And then, we can calculate the concentration of the analyte in our sample solution. And such equation which links together the output quantity with all the input quantities is called measurement model. So, this is here a typical measurement model for a titration with 1 to 1 stoichiometry. So, we have here the volume of titrant that was consumed, the molar concentration of the titrant and then the volume of the sample solution which was taken for titration. And from this data we can calculate the analyte concentration in the sample solution. And now, it's important to ask that if we know the uncertainties of the input quantities and if we know what the model looks like. How do we find the combined standard uncertainty of the output quantities ? And there are several simpler cases and one general case. So, first of all, the simplest case that can be, is that all the factors or all the input quantities are simply summed or subtracted to get, the output quantity value. A typical case where this sort of equation holds is the volume of a pipette where the different uncertainty sources can be considered as factors or input quantities and their uncertainties are then combined according to this simple rule which we have already seen. So, if our equation contains only additions and subtractions, we simply take all the standard uncertainties of all the input quantities, take them to the square, sum them up and take square root. It's important to note that even though we have here subtraction and here addition, we nevertheless always write additions here. So it does not matter whether it is plus or minus, here it will always be plus. And of course it's easy to follow that this is correct way of doing, that all these quantities of course need to have the same units. Only then it's allowed to combine the standard uncertainties as this. And, of course, it does once again remember that all the uncertainties, before we combine them, need to be converted to the standard uncertainty. Now the second very common type of measurement model is model that is composed only of multiplications and divisions and, in fact, the titration model that we saw a few slides ago is a typical example of this. In this case now, the combined uncertainty, the combined standard uncertainty of the output quantity is calculated in a slightly more complex way. Now we do not add combined standard uncertainties of the input quantity because very often those input quantities, in fact, have different units, so, we are not allowed to add. Instead, we add their relative standard uncertainties. So, in all these cases the standard uncertainty of the respective input quantity is divided by its value, standard uncertainty divided by value, etc. And again importantly, whether it is multiplication or division, we always have "+" signs here we never put "-" into this equation. And, when this here is calculated, then this expression actually gives us the relative combined standard uncertainty of the output quantity. And, this relative combined standard uncertainty of the output quantity can be multiplied by the value of the output quantity. So that we can get the combined standard uncertainty of the output quantity in the same unit as this output quantity is measured. And, now the general case, the output quantity is now found as a function of the input quantities whereby it is not important what mathematical operations are included here. They can be summing, subtracting, multiplications, divisions, squares, square roots, logarithms, etc. And, in this general case, the combined standard uncertainty of the output quantity is found from square, the square root of squared summing of such uncertainty components. So, each of these terms here is called uncertainty component of the respective input quantity. And, each of them, in turn, is composed of the standard uncertainty of the input quantity and the partial derivative of the output quantity with respect to that input quantity. In fact, the two previous cases are special cases from this general case. So, if the partial derivative calculation is done, then, for those previous cases, we arrive at those more simple equations. But, this equation is completely universal. And, we will use it later in this course, and, I will also demonstrate for you how you can calculate those partial derivatives quite easily without going very deeply into mathematics. And, also it is important to stress that all those equations on the previous slides, they strictly saying, work only for non correlated input quantities. And, in most of the cases in chemical analysis, we can regard input quantities as non correlated. But, there will be few special cases which I will then demonstrate separately where they release correlation. But, at least, in simple chemical analysis, correlation usually does not have such a strong effect that we would really need to worry about it very much. |
Examinons maintenant le concept clé d'estimation de l'incertitude de mesure: l’incertitude-type composée. Si nous parlons de mesures, les résultats de mesure sont presque toujours influencés par différents facteurs ou ils peuvent être calculés à partir de différents paramètres ou quantités. Et ainsi, comme on l'appelle dans la science de la mesure, notre résultat de mesure est une quantité de sortie qui est influencée par, ou calculée à partir, d'un certain nombre de quantités d'entrée. Et maintenant, l'incertitude-type composée est l'incertitude de la quantité de sortie qui prend en compte les incertitudes de toutes les quantités d'entrée. Et l'incertitude-type composée comme nous l'avons déjà vu dans le chapitre précédent est indiquée par uc, avec C signifiant composée. Et la plupart des mesures en chimie sont des mesures indirectes, ce qui signifie que nous ne voyons pas afficher immédiatement le résultat sur notre instrument de mesure mais plutôt que nous mesurons différentes quantités et différents paramètres et calculons à partir de leurs valeurs notre mesure résultante. Et toutes ces mesures proviennent des valeurs des quantités d'entrée et les quantités de sortie calculées sont appelées mesures indirectes. Et le bon exemple est le titrage. Lorsque nous effectuons le titrage, nous devons connaître la concentration de titrant, nous devons connaître le volume de notre solution d'échantillon que nous avons prise pour le titrage et nous devons également connaître le volume de titrant qui a été consommé pour ce titrage. Et ensuite, nous pouvons calculer la concentration de l'analyte dans notre solution échantillon. Et une telle équation qui relie la quantité de sortie avec toutes les quantités d'entrée est appelée modèle de mesure. Voici donc un modèle de mesure typique pour un titrage avec une stœchiométrie de 1 pour 1. Donc, nous avons ici le volume de titrant qui a été consommé, la concentration molaire du titrant et puis le volume de la solution d'échantillon qui a été prélevée pour le titrage. Et à partir de ces données, nous pouvons calculer la concentration d'analyte dans la solution d'échantillon. Et maintenant, il est important de demander que si nous connaissons les incertitudes des quantités d'entrée et si nous savons à quoi ressemble le modèle : comment trouver l'incertitude type composée de la quantité de sortie ? Il existe plusieurs cas plus simples et un cas général. Donc, tout d'abord, le cas le plus simple qui puisse être, est que tous les facteurs ou toutes les quantités d'entrée sont simplement additionnés ou soustraits pour obtenir la valeur de la quantité de sortie. Un cas typique où ce type d'équation est valable est le volume d'une pipette où les différentes sources d'incertitude peuvent être considérées comme des facteurs ou des quantités d'entrée et leurs incertitudes sont ensuite combinées selon cette règle simple que nous avons déjà vue. Donc, si notre équation ne contient que des additions et des soustractions, nous prenons simplement toutes les incertitudes types de toutes les quantités d'entrée, nous les élevons au carré, les additionnons et nous prenons la racine carrée. Il est important de noter que même si nous avons ici la soustraction et ici l'addition, nous écrivons néanmoins toujours des additions ici. Donc, peu importe que ce soit plus ou moins, ici ce sera toujours plus. Et bien sûr, il est facile de comprendre que c'est la bonne façon de faire, que toutes ces quantités doivent bien sûr avoir les mêmes unités. Ce n'est qu’après cela qu'il est permis de combiner les incertitudes types comme ceci. Et, bien sûr, on se souvient encore une fois que toutes les incertitudes, avant de les combiner, doivent être converties en incertitude-type. Maintenant, le second type de modèle de mesure très courant est un modèle qui est seulement composé de multiplications et de divisions, et, en fait, le modèle de titrage que nous avons vu dans les diapositives précédentes, en est un exemple typique. Dans ce cas, l’incertitude composée, l'incertitude-type composée de la quantité de sortie est calculée d'une manière légèrement plus complexe. Nous n'ajoutons pas les incertitude-types composées de la quantité d'entrée parce que, très souvent, ces quantités d'entrées ont, en fait, différentes unités, nous ne sommes donc pas autorisés à les ajouter. A la place, nous ajoutons leur incertitude-type relative. Ainsi, dans tous ces cas, l'incertitude-type de chaque quantité d'entrée respective est divisée par sa valeur, l'incertitude-type divisée par sa valeur, etc. A nouveau ce qui est important, qu'il s'agisse d'une multiplication ou d'une division, est que nous avons toujours ici des signes "+", nous ne mettons jamais des "-" dans cette équation. Et, lorsque cela est calculé, alors cette expression nous donne, en fait, l'incertitude-type composée relative de la quantité de sortie. Cette incertitude-type composée relative de la quantité de sortie peut être multipliée par la valeur de la quantité de sortie afin que nous puissions avoir l'incertitude-type composée de la quantité de sortie dans la même unité que celle dans laquelle la quantité de sortie est mesurée. Et, maintenant, le cas général, la quantité de sortie est maintenant trouvée comme une fonction des quantités d'entrées, selon laquelle il n'est pas important de savoir quelles opérations mathématiques sont incluses ici. Il peut s'agir d'additions, de soustractions, de multiplications, divisions, de carrés, de racines carrées, de logarithmes, etc. Et, dans ce cas général, l'incertitude-type composée de la quantité de sortie est trouvée à partir du carré, la racine carrée de la somme au carrée de telles composantes de l'incertitude. Ainsi, chacun de ces termes est appelé composante d'incertitude de la quantité d'entrée respective. Et, chacune d'entre elles est, à son tour, composée de l'incertitude-type de la quantité d'entrée et de la dérivée partielle de la quantité de sortie par rapport à cette quantité d'entrée. En fait, les deux cas précédents sont des cas particuliers de ce cas général. Ainsi, si le calcul de la dérivée partielle est effectué, alors, pour ces cas précédents, nous arrivons à ces équations plus simples. Mais, cette équation est complètement universelle. Et, nous l'utiliserons plus tard, et, je vous démontrerai également comment vous pouvez calculer ces dérivées partielles assez facilement sans aller très profondément dans les mathématiques. Et, il est également important de souligner que toutes ces équations sur les diapositives précédentes, à proprement parler, ne fonctionnent que pour les entrées non corrélées. Et, dans la plupart des cas pour les analyses chimiques, nous pouvons considérer les quantités d'entrées comme non corrélées. Mais, il y aura peu de cas particuliers que je démontrerai ensuite séparément où ils libèrent une corrélation. Mais, au moins, dans l'analyse chimique simple, la corrélation n'a généralement pas un effet si fort que nous aurions vraiment besoin de beaucoup nous en inquiéter. |
Une mesure indirecte est une mesure dans laquelle la quantité en sortie (résultat) est obtenue par un calcul (en utilisant une équation du modèle) à partir de plusieurs quantités en entrée. Un exemple typique est le titrage. En cas de titrage avec un rapport molaire de 1: 1, la concentration en analyte dans la solution d'échantillon CS (la quantité de sortie) est exprimée par les quantités d'entrée - volume de solution d'échantillon prélevé pour titrage (VS), concentration en titrant (CT) et volume de titrant consommé. pour le titrage (VT) - de la façon suivante :
(4.5)
Dans le cas général, si la quantité en sortie Y est trouvée à partir des quantités en entrée X1, X2, … Xn selon une fonction F
(4.6)
alors, l'incertitude type composée de la quantité de sortie uc(y) peut être exprimée via les incertitudes-types des quantités d'entrée u(xi) de la façon suivante :
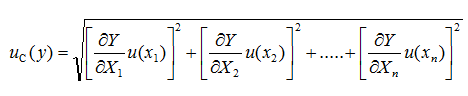 (4.7)
(4.7)
Les termes 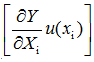 sont les composantes de l'incertitude. Les termes
sont les composantes de l'incertitude. Les termes 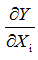 sont des dérivées partielles. À première vue, l'eq 4.7 peut sembler très complexe mais elle n'est en fait pas trop difficile à utiliser : les composantes de l'incertitude peuvent être calculées numériquement en utilisant la méthode du tableur de Kragten (comme le montre le chapitre 9.7).
sont des dérivées partielles. À première vue, l'eq 4.7 peut sembler très complexe mais elle n'est en fait pas trop difficile à utiliser : les composantes de l'incertitude peuvent être calculées numériquement en utilisant la méthode du tableur de Kragten (comme le montre le chapitre 9.7).
Dans des cas spécifiques, des équations plus simples sont valables. Si la quantité de sortie est exprimée via les quantités d'entrée comme suit :
image perdue (4.8)
alors :
image perdue (4.9)
De manière importante, que les quantités d'entrées soient ajoutées ou soustraites, les incertitudes types au carré sous la racine carrée sont toujours ajoutées. Cette manière de combiner les composantes d'incertitude est la même que celle utilisée ci-dessus pour le pipetage.
Si le modèle de mesure est
image perdue (4.10)
image perdue (4.11)
Comme on peut le constater, ce sont ici les incertitudes-types relatives qui sont combinées et la somme au carré nous donne l’incertitude-type relative composée de la quantité produite. L’incertitude-type composée absolue de la quantité obtenue est la suivante:
image perdue (https://sisu.ut.ee/sites/default/files/styles/os_files_xxlarge/public/measurement/files/valem42-492.png?itok=fpv7u7EA)(4.12)
Autoévaluation sur cette partie du cours : Test 4.2
https://sisu.ut.ee/measurement/measurement-1-9
5.3. Comparaison des composants de l'incertitude
Résumé : Les composantes d’incertitude du chapitre précédent sont comparées. La propriété de la sommation au carré - supprimant les composantes d'incertitude les moins influentes - est expliquée. La signification de l'estimation de l'incertitude type composée obtenue est expliquée en termes de probabilité (la probabilité que la valeur réelle du volume pipeté se situe dans la plage d'incertitude calculée).
Comparaison des composantes de l'incertitude
http://www.uttv.ee/naita?id=17578
|
Let us now examine a little bit closer those values and what do we see. If we would simply sum up these three values, we would get altogether 0.028. However what we see here, we have only 0.019. So we see that actually, the combined standard uncertainty is quite similar by its magnitude to one of the uncertainty components and this uncertainty components as we easily can see is the largest of the three. This is the uncertainty due to the calibration and this is now the general property of this square type of summing. Whenever you sum quantities of different magnitude, then the result will be often quite similar to the biggest one, meaning the biggest one will dominate in the result, suppressing the smaller ones. This is also the reason why if there are some very small uncertainty components, they wouldn't even have any influence at all. So suppose we would have some 0.001 mL uncertainty components here, it would be fairly safe to neglect them because they wouldn't influence the combined standard uncertainty at all. In the case of uncertainty modeling and initial calculations, usually all uncertainty sources that are meaningful are considered and they are quantified because beforehand we don't yet know which of them are important and which of them are not so important. We have now managed to complete our first uncertainty calculation and it is now interesting to think: what does this uncertainty tell us? This uncertainty tells us the following: if we now do more pipetting with the same pipette, then the volumes that we get with this pipetting are in the following region. In this region, with the probability of roughly 68%. So every time we quantify our uncertainty as standard uncertainty, we always have the 68% probability. |
Examinons maintenant d'un peu plus près ces valeurs et ce que nous observons. Si nous additionnons simplement ces trois valeurs, nous obtenons purement et simplement 0,028. Toutefois, nous voyons ici que nous n'avons que 0,019. Donc on voit en fait que l'incertitude-type composée est quasiment similaire par son ampleur à l’une des composantes de l'incertitude et cette composante de l'incertitude comme nous pouvons le voir facilement est la plus grande des trois. C'est l'incertitude due à la calibration et c'est maintenant la propriété générale de cette somme des carrés. A chaque fois que vous sommez des quantités d'ampleur différentes, alors le résultat sera souvent similaire à la plus grande valeur, ce qui signifie que la plus grande dominera dans le résultat, supprimant les plus petites valeurs. C'est donc la raison pour laquelle s'il y a de très petites composantes de l'incertitude, elles n'auront pas du tout d'influence. Supposons que nous avons 0,001 mL comme composantes d'incertitude ici, il serait plus prudent de les négliger car elles n'influenceront pas l'incertitude-type composée du tout. Dans le cas de la modélisation de l'incertitude et de calculs initiaux, généralement toutes les sources d'incertitudes qui sont significatives sont considérées et elles sont quantifiées car nous ne savions pas au préalable lesquelles étaient importantes et lesquelles n'étaient pas importantes. Nous avons maintenant réussi à terminer notre premier calcul d'incertitude et maintenant, il a une chose intéressante : que nous dit cette incertitude ? Cette incertitude nous dit la chose suivante : si nous procédons à plus de pipetages avec la même pipette, alors les volumes que nous obtenons avec ce pipetage se situent dans la région suivante. Dans cette région, avec la probabilité d'à peu près 68%. Donc à chaque fois qu'on quantifie notre incertitude en tant qu'incertitude type, on aura toujours une probabilité de 68%. |
Nous pouvons voir que la composante d'incertitude avec la plus grande magnitude est l'incertitude d'étalonnage u (V, CAL) = 0,017 mL. L’incertitude type composée est du même ordre de grandeur : uc (V) = 0,019 mL. Si la somme était établie non pas par un calcul de la racine carrée de la somme des carrés, mais par une simple somme arithmétique, la valeur serait 0,028 mL. C'est une bonne illustration de la propriété de la somme des carrés : les composantes d'incertitude les plus petites sont minimisées par les composantes d'incertitude les plus grandes.
L’idée de la sommation au carré des composantes est que les différents effets générateurs d’incertitude influent sur le résultat dans différentes directions (s'annulant ainsi partiellement) et que leurs amplitudes ne sont pas nécessairement égales aux valeurs des estimations de l’incertitude, mais peuvent aussi être plus petites (voir chapitre 1).
Examiner les contributions à l’incertitude est très utile si l’on veut réduire l’incertitude. Afin de réduire l'incertitude d'une mesure particulière, il est toujours nécessaire de s'attacher à réduire les incertitudes causées par les composantes les plus importantes. Donc, dans ce cas, il n'est pas très utile d'acheter un climatiseur plus coûteux pour la pièce car l'amélioration de l'incertitude qui en résultera sera faible. Il sera également impossible d'améliorer nettement l'incertitude en réduisant la composante de répétabilité. Il est clair que quoi que l’on fasse avec ces deux composantes, l’incertitude type composée uc (V) (équation 4.4) ne peut pas passer de 0,019 mL à moins de 0,017 mL, ce qui n’est pas une diminution significative. Ainsi, si un pipetage plus exact est nécessaire, la meilleure chose à faire consiste à calibrer la pipette en laboratoire. De cette manière, il est réaliste d’obtenir une incertitude d’étalonnage trois fois plus basse, ce qui conduit à une incertitude composée deux fois plus faible du volume à la pipette. Voir la section 4.6 pour un exemple.
Autoévaluation sur cette partie du cours : Test 4.3
https://sisu.ut.ee/measurement/measurement-1
5.4. Incertitude élargie
https://sisu.ut.ee/measurement/44-expanded-uncertainty
Résumé : La probabilité d'environ 68% fournie par l'incertitude-typ est souvent trop faible pour les utilisateurs de l'incertitude de mesure. Par conséquent, l'incertitude de mesure est présentée aux clients principalement comme une incertitude élargie, U. L'incertitude élargie est calculée à partir de l'incertitude-type en la multipliant par un facteur d'élargissement, k.
Dans le cas de l'exemple de pipetage, l'incertitude élargie à k=2 se présente comme suit :
U (V ) = uc (V ) · k = 0.019 mL · 2 = 0.038 mL (4.13)
L’incertitude élargie au niveau k=2 est le moyen le plus courant d’exprimer l’incertitude des résultats de mesure et d’analyse.
Calcul de l'incertitude élargie
http://www.uttv.ee/naita?id=17579
| Now even though standard uncertainty is very good and useful for uncertainty calculations, for those people who actually use measurement and analysis results, the customers, 68% often is too low probability. Because it essentially means that every third time you pipette, it can be outside that range, or if some customer gets different analysis results, that every third result actually is such that the true value is outside the uncertainty range. Therefore uncertainty results are reported usually not as standard uncertainties, they usually report it as expanded uncertainties. And now if we remember the normal distribution then in the normal distribution curve, if we multiply standard deviation by 2 we get the probability of 95% and if we multiply the standard deviation by three we get the probability of higher than 99%. And this possibility also is used here with the uncertainty estimation. So very often, results are presented with the expanded uncertainties and the expanded uncertainties are denoted by capital "U", so standard uncertainty always is denoted by small "u" but by the capital "U" we denote expanded uncertainty. And expanded uncertainty is found by multiplying standard uncertainty with the so called coverage factor. Coverage factor is denoted by small "k". Coverage factor in principle can have any values but most usually it's either 2 or 3 or between 2 and 3. And out of those possibilities 2 is by far the most common. Let us see how we could present our uncertainty as expanded uncertainty and let us pick coverage factor 2. It is equal to 0.019 multiplied by 2, which is equal to 0.038 mL. And this 2 is coverage factor. | Maintenant, même si l'incertitude-type est très bonne et utile pour les calculs d'incertitude, pour les personnes qui utilisent réellement des résultats de mesure et d'analyse, les clients, 68% est souvent une probabilité trop faible. Parce que cela signifie essentiellement qu'au bout de trois fois que vous pipetez, il peut être en dehors de cette plage, ou si un client obtient des résultats d'analyse différents, que chaque troisième résultat est tel que la vraie valeur est en dehors de la plage d'incertitude. Par conséquent, les résultats d'incertitude ne sont pas signalés généralement comme une incertitude-type, ils les signalent généralement comme des incertitudes élargies. Et maintenant si on se rappelle de la distribution normale alors dans la courbe de distribution normale, si on multiplie l'écart-type par 2 nous obtenons une probabilité de 95% et si on multiplie l'écart type par trois nous obtenons une probabilité supérieure à 99%. Et cette possibilité est également utilisée ici avec l'estimation de l'incertitude. Très souvent, les résultats sont présentés avec les incertitudes élargies et les incertitudes étendues sont dénotées par la lettre majuscule "U", donc l'incertitude-type est toujours désignée par un petit "u" mais par le "U" majuscule nous dénommons une incertitude élargie. Et l'incertitude élargie se trouve en multipliant l'incertitude-type avec le dit facteur d'élargissement. Le facteur d'élargissement est indiqué par un petit "k". En principe, le facteur d'élargissement peut prendre toutes les valeurs, mais le plus souvent c'est soit 2 ou 3 soit entre 2 et 3. Et entre ces possibilités, 2 est la plus commune. Regardons ensemble comment nous pouvons présenter nos incertitudes comme incertitude élargie et prenons 2 comme facteur d'élargissement. C'est égal à 0,019 multiplié par 2, qui est égal à 0,038 mL. Et ce 2 est le facteur d'élargissement. |
Autoévaluation sur cette partie du cours : Test 4.4
5.5. Présentation des résultats de mesure
https://sisu.ut.ee/measurement/45-presenting-measurement-results
Résumé : Le résultat du pipetage - la valeur et l’incertitude élargie - est présenté. Il est important de préciser clairement ce qui a été mesuré. La présentation correcte du résultat de mesure comprend la valeur, l’incertitude et des informations sur la probabilité de l’incertitude. Il est expliqué qu'en termes simplifiés, nous pouvons supposer que k = 2 correspond à environ 95% de la probabilité de couverture. Il est expliqué comment décider du nombre de décimales à donner lors de la présentation d’un résultat de mesure et de l’incertitude.
La présentation correcte du résultat de la mesure dans ce cas se présenterait comme suit:
Le volume du liquide pipetté est V = (10.000 ± 0.038) mL, k = 2, norm. (4.14)
Les parenthèses signifient que l’unité «mL» est valable à la fois pour la valeur et pour l’incertitude. «norm.» signifie que la quantité de sortie devrait être approximativement normalement distribuée. Ceci, associé à la valeur 2 du facteur d'élargissement, signifie que l'incertitude présentée devrait correspondre à une probabilité de couverture d'environ 95% (voir le chapitre 3.1 pour plus de détails).
Quand pouvons-nous supposer que la quantité de sortie est normalement distribuée? C'est-à-dire, quand pouvons-nous écrire «norm» en plus du facteur d'élargissement ? Il n’est pas simple de répondre rigoureusement à cette question, mais une règle simple est que, s’il existe au moins trois sources principales d’incertitude ayant une influence comparable (c’est-à-dire que les composantes d’incertitude les plus petites et les plus grandes diffèrent d'un facteur 3 ou moins), nous pouvons supposer que la fonction de distribution de la sortie sera suffisamment similaire à une distribution normale. [1]
Le chapitre 9.8 présente une approche plus sophistiquée du calcul de l’incertitude élargie qui correspond à une probabilité de couverture concrète.
Présentation des résultats de mesure
http://www.uttv.ee/naita?id=17576
| We managed to complete our first uncertainty calculation and also we managed to find the expanded uncertainty. And suppose now that we want to present this volume with its uncertainty as a result to somebody who is interested in this measurement. So let us see how to correctly present the measurement or an analysis result, so the presentation would be something as follows. First of all we need to clearly say what it is that we actually measured. So, the volume of the liquid that we pipetted this is our measurement, this is what we measure, we denote it by V it is equal to, and our nominal volume of the pipette that we use was 10 mL, our uncertainty, and here we put it as expanded uncertainty, 0.038 mL we now close the brackets and write the unit. These brackets are needed to denote that the unit goes both for the uncertainty and for this 10 also so that instead of writing milliliters two times people use brackets and put milliliters behind there. But now this is not yet enough you remember that uncertainty always is associated with some kind of probability, so we never can guarantee that the pipetted volume really is with 100% probability within this range. Therefore we also must give some information to the people about the probability distribution. And in this particular case we can write the following. K is equal to two and norm and what does this mean this means that we have calculated the combined standard uncertainty, multiplied by the coverage factor, 2, obtain this expanded uncertainty and it means that we have a reason to expect that our measurement obeys the normal distribution and if both of these are so, then according to the properties of the normal distribution the person who uses this result can assume that the probability of the true value being within this range is roughly 95%. This is simplified and we will see later on that it is oftentimes more complex than this but in the simple cases this is a fully valid and useful approach. Whenever we present the result, we also have to think about the number of decimals that we give here. I've put here three decimals both for the result and for the uncertainty but I did not explain in any way why I picked this number of decimals. There are several conventions that I used so that there is no one single universal rule but I will give you one approach that is fairly reasonable and usable in almost all cases. So whenever you want to decide how many decimals after the comma you have to give. You first start with the uncertainty, so step one is to look at the uncertainty. And with the uncertainty you will have to look at the number of significant digits and significant digit means the following, you start counting the digits from the left and any digit that is not 0 is called significant digit. So we come here, 0, not the significant digits, 0, not the significant digit, 3 is a significant digit and 8 also so we get 1, 2 digits. So we have here 2 significant digits, and the general rule goes as follows : if the first significant digit is between 1 and 4 ; in the range 1 to 4 then we give 2 significant digits in the uncertainty. 1, 2 ; but if the first significant digit is the range of 5 to 9, in this case we give 1 significant digit. So, and once we have managed to find how many digits we give with the uncertainty and here it turns out that we give 2 significant digits which in turn, in this particular case, means that we give 1, 2, 3 decimals or three digits after the comma, when we have sorted this out then we take the next step and we look at the value. The value, and here the rule is very simple for the value we give just as many decimals after the comma as the uncertainty has. Easy. | Nous avons réussi à compléter notre premier calcul d'incertitude et aussi nous avons trouvé l'incertitude élargie. Et imaginez maintenant que nous voulons présenter ce volume avec son incertitude comme résultat à quelqu'un qui est intéressé par cette mesure. Alors regardons comment présenter correctement la mesure ou un résultat d'analyse, alors la présentation serait quelque chose comme ce qui suit Tout d'abord nous devons clairement dire ce qu'est vraiment ce que nous avons mesuré. Alors, le volume de liquide que nous avons pipeté c'est notre mesure, c'est ce qu'on mesure, nous l'indiquons par V qui est égal à, et notre volume nominal de la pipette utilisée est 10 mL, notre incertitude, et ici nous la mettons comme l'incertitude élargie, 0,038 mL nous fermons maintenant les parenthèses et nous écrivons l'unité. Les parenthèses sont nécessaires pour indiquer que l'unité va pour les deux pour l'incertitudes et pour ce 10 aussi afin de ne pas écrire millilitre deux fois on utilise des parenthèses et on met millilitre derrière elle. Mais maintenant ce n'est pas encore assez, vous vous souvenez que l'incertitude est toujours associée avec une sorte de probabilité, donc nous ne pouvons jamais garantir que le volume pipeté est vraiment avec 100% de probabilité dans la plage. Par conséquent nous devons donner quelques informations aux personnes concernant la loi de probabilité. Et dans ce cas particulier nous pouvons écrire ce qui suit. K est égal à 2 et norm et qu'est-ce que cela signifie, cela signifie que nous avons calculé l'incertitude-type composée, multiplié par le facteur d'élargissement, 2, obtenu cette incertitude élargie et cela signifie que nous avons une raison d'espérer que notre mesure obéisse à la distribution normal et si ces deux le sont, alors selon les propriétés de la distribution normale la personne qui utilise ce résultat peut supposer que la probabilité d'avoir la vraie valeur dans cette plage est approximativement 95%. Ceci est simplifié et nous verrons plus tard que c'est très souvent plus complexe que cela mais dans les cas simples c'est une approche complètement valide et utile. Chaque fois que nous présentons le résultat, nous devons aussi penser aux nombres de décimales que nous avons ici. J'ai ici mis 3 décimales pour le résultat et pour l'incertitudes mais je n'ai expliqué d'aucune manière pourquoi j'ai choisi ce nombre de décimales. Il y a plusieurs conventions que j'ai utilisé pour qu'il n'y ait aucune règle universelle mais je vais vous donner une approche qui est plutôt raisonnable et utilisable dans presque tous les cas. Donc chaque fois que vous voudrez décider combien de décimales après la virgule vous devez mettre. Premièrement commencez avec l'incertitude, donc la première étape est de regarder l'incertitude. Et avec l'incertitude vous devrez regarder au nombre de chiffres significatifs et chiffre significatif signifie ce qui suit, vous commencez à compter les chiffres par la gauche et tout chiffre qui n'est pas 0 est appelé chiffre significatif. Alors nous allons ici, 0, pas un chiffre significatif, 0, pas un chiffre significatif, 3 est un chiffre significatif et 8 l'est aussi alors nous avons 1, 2 chiffres. Alors nous avons ici 2 chiffres significatifs, et la règle générale est comme suit : si le premier chiffre significatif est entre 1 and 4 ; entre 1 et 4 alors nous avons 2 chiffres significatifs dans l'incertitude. 1, 2 ; mais si le premier chiffre significatif est entre 5 et 9, dans ce cas nous donnons 1 chiffre significatif. Alors, et une fois que nous avons réussi à trouver combien de chiffres nous donnons avec l'incertitude et ici il s'avère que nous donnons 2 chiffres significatifs qui à leur tour, dans ce cas particulier, signifient que nous donnons 1, 2, 3 décimales après la virgule, quand nous avons réglé ça nous commençons l'étape suivante et nous regardons la valeur. La valeur, et ici la règle est très simple pour la valeur nous donnons autant de décimales après la virgule que l'incertitude a. Facile. |
Autoévaluation sur cette partie du cours Test 4.5
https://sisu.ut.ee/measurement/measurement-1-2
***
[1] Dans ce cas particulier, il existe une quantité dominante avec une distribution supposée rectangulaire, ce qui conduit à une fonction de distribution avec des "queues très faibles" (ce qui signifie: ceci n'est en fait pas exactement une distribution normale). Ainsi, une probabilité de couverture de 95% est déjà atteinte avec une incertitude élargie de 0,0034 mL (comme en témoignent les simulations de Monte Carlo). Ainsi, l’incertitude présentée de 0,0038 mL est une estimation prudente (ce qui n’est pas mauvais comme expliqué au chapitre 3.5).
5.6. Exemple pratique
https://sisu.ut.ee/measurement/41-n%C3%A4idis%C3%BClesandeks
Ceci est un exemple de calcul du volume de liquide délivré et de son incertitude à partir d’une pipette volumétrique auto-étalonnée.
L'incertitude du volume pipeté u (V) comporte trois composantes principales: l'incertitude due à la répétabilité, u (V, rep); incertitude due à l'étalonnage de la pipette, u (V, cal) et incertitude liée à la différence entre la température et 20°C, u (V, temp).
L'estimation de la différence maximale probable du volume de la pipette par rapport au volume nominal, exprimée sous la forme ± x, est souvent utilisée comme estimation de l'incertitude d'étalonnage de la pipette (comme indiqué au chapitre 4.1). Il est généralement donné par le fabricant sans aucune information supplémentaire sur sa probabilité de couverture ou sa fonction de distribution. Dans un tel cas, il est le plus sûr de supposer que la distribution rectangulaire est valide et de convertir l'estimation de l'incertitude en incertitude-type en la divisant par la racine carrée de trois.
Habituellement, dans le cas d'un travail de haute exactitude, la pipette est étalonnée en laboratoire afin d'obtenir une incertitude d'étalonnage inférieure. Comme on l’a vu dans les chapitres 4.2 et 4.3, si l’incertitude due à l’étalonnage en usine est utilisée, cette composante de l’incertitude d’étalonnage est la plus influente. Donc, le réduire réduirait également l'incertitude globale. Il est très important que l'étalonnage et le pipetage soient effectués dans les mêmes conditions et de préférence par la même personne. A partir des données de calibration, nous pouvons obtenir deux informations importantes: (1) le terme de correction pour le volume de pipette V correction avec incertitude u (V, cal) et (2) la répétabilité de pipetage u (V, rep). L'exemple présenté ici explique ces points.
Pour étalonner une pipette, on pipette plusieurs fois de l'eau (à une température contrôlée afin de connaître sa densité), les masses des quantités d'eau pipetées sont mesurées et les volumes d'eau pipetés sont calculés (en utilisant la densité de l'eau à la température d'étalonnage). Voici les données d'étalonnage d'une pipette de 10 mL:
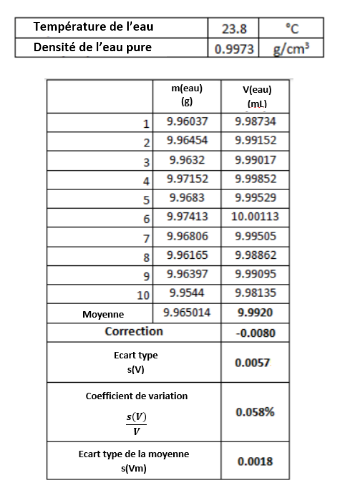
L’écart type est calculé selon l’équation:
image perdue (4.15)
L'incertitude due à la répétabilité du pipetage u (V, REP) est égale à cet écart-type de 0,0057 ml. Le pipetage est souvent utilisé dans les analyses de titrage. Si la solution titrée est pipetée, cette contribution à la répétabilité est déjà prise en compte dans la répétabilité des résultats du titrage et n'est pas prise en compte séparément dans l'incertitude du volume de la pipette.
L'incertitude de l'étalonnage (en fait, l'incertitude de la correction) doit toujours être prise en compte. Dans de nombreux cas, l’incertitude liée à la répétabilité de l’obtention de la correction est la seule source d’incertitude importante et les autres sources peuvent être exclues . Cette incertitude est exprimée (lorsque l'étalonnage est effectué [1]) en tant qu'écart type de la moyenne :
image perdue (4.16)
Dans cet exemple, la correction est de -0,0080 mL et son incertitude-type est de 0,0018 mL.
Lorsqu'il existe une possibilité que le pipetage soit effectué à une température différente de celle du calibrage (et que cette possibilité existe presque toujours), une source d'incertitude supplémentaire due au changement de température est introduite et doit être prise en compte.
Dans ce cas, nous supposons que la température de la pipette ne diffère pas de la température d’étalonnage de plus de 4°C (Δt = ± 4°C, en supposant une distribution rectangulaire). La densité de l’eau dépend de la température; nous devons donc également prendre en compte le coefficient de dilatation thermique de l’eau, qui est γw = 2,1 · 10-4 1/°C. Alors,
image perdue (4.17)
Maintenant, lorsque nous allons effectuer un seul pipetage, le volume est de 9,992 ml et son incertitude-type composée est
image perdue (4.18)
L'incertitude élargie k = 2 [2] du volume à la pipette peut être trouvée comme suit:
U(V ) = uc(V ) · k = 0.0077 · 2 = 0.0154 mL (4.19)
Comme expliqué à la section 4.5, si le premier chiffre significatif de l'incertitude est compris entre 1 et 4, il convient de présenter l'incertitude avec deux chiffres significatifs. Ainsi, nous pouvons écrire le résultat:
Le volume du liquide pipeté est:
V = (9.992 ± 0.015) mL, k = 2, norm. (4.20)
Il est intéressant de comparer maintenant cette incertitude élargie à l’incertitude élargie obtenue au chapitre 4.5 (équation 4.14). Nous constatons que lorsque la pipette est étalonnée dans notre laboratoire, l'incertitude du volume est plus de deux fois inférieure. Nous voyons également que la composante d'incertitude due à l'étalonnage de la pipette, qui était à l'époque la plus grande composante d'incertitude, est maintenant la plus petite.
***
[1] Lorsque l'étalonnage n'est pas effectué en laboratoire, l'incertitude de l'étalonnage peut être prise en compte conformément aux informations manuelles, qui sont généralement indiquées sur la pipette en tant que plage de tolérance. Par exemple, si le fabricant a fourni la plage de tolérance de ± 0,03 mL (dans le cas d’une pipette de 10 mL), l’incertitude-type de la correction de la pipette est calculée comme suit [image perdue]. La valeur de correction dans ce cas est de 0,00 mL.
[2] Nous verrons plus loin dans ce cours (chapitre 9.8) comment trouver rigoureusement si nous pouvons dire que l’incertitude élargie de k = 2 dans un cas particulier correspond à 95% (cela dépend du nombre effectif de degrés de liberté). Et sinon, quel k devrait être utilisé pour atteindre une probabilité de couverture d’environ 95%.
Dans cet exemple, le nombre effectif de degrés de liberté est de 26 et le facteur de couverture respectif (en réalité, le coefficient de Student) avec une probabilité de 95% est de 2,06, ce qui n’est que très légèrement différent de 2 (l’incertitude accrue augmenterait de 0,015 mL à 0,016 mL).
6. Principe de l'estimation de l'incertitude de mesure
https://sisu.ut.ee/measurement/5-sources-measurement-uncertainty-chemical-analysis
Résumé : Les exemples de détermination des pesticides dans les oranges présentent les principes fondamentaux de l’estimation de l’incertitude de mesure, appelés principes de GUM. Ces principes ont été énoncés dans l'ISO GUM [1] et sont désormais universellement reconnus comme le fondement commun de toutes les différentes méthodes d'estimation de l'incertitude.
Ces principes sont les suivants:
- La base de toute mesure (donc évidemment aussi l'évaluation de l'incertitude de mesure) est la définition du mesurande;
- La procédure de mesure utilisée doit correspondre à la définition du mesurande;
- Toutes les sources d'incertitude pertinentes doivent être soigneusement examinées et celles qui sont importantes doivent être prises en compte;
- Les effets aléatoires et systématiques sont traités de la même manière lors de l'estimation de l'incertitude de mesure - les deux sont évalués en tant qu'incertitudes-types, qui sont ensuite combinées dans l'incertitude-type composée.
***
[1] ISO GUM se réfère à l'origine au Guide pour l'expression de l'incertitude de mesure, ISO, Genève, Suisse, 1993 (réimprimé en 1995). En 2008, ce document a été révisé et réédité sous le titre ISO JCGM 100: 2008 Évaluation des données de mesure - Guide pour l'expression de l'incertitude de mesure. Ce dernier document est disponible en ligne à l'adresse suivante : https://www.bipm.org/fr/publications/guides/gum.html
Déposé sur cette espace 10/2019 version anglaise : JCGM_100_GUM95rev2008_E.pdf
Déposé sur cette espace 10/2019 version française : JCGM_100_GUM95rev2008_F.pdf
6.1. Définition du mesurande
https://sisu.ut.ee/measurement/51-measurand-definition
Résumé : Le premier principe de l’incertitude de mesure est le suivant: le mesurande doit être défini correctement et sans ambiguïté. L'importance de la définition des mesurandes est expliquée dans l'exemple de la détermination de la teneur en pesticide dans les oranges.
Définition du mesurande
http://www.uttv.ee/naita?id=17585
| We will now look at certain basic principles of measurement uncertainty estimation. And the principle number one, is measurand definition. This is an extremely important thing which sometimes is slightly overlooked. But I'm going to explain why it is extremely important to very clearly define what the measurand is and I'm going to do that on an example. And let us take this orange as our example, and suppose our aim, our purpose of the analysis, is determination whether this orange is suitable for eating based on its pesticide content. Meaning whether there are small enough concentrations of pesticides in this orange. Nowadays unless it's a purely organically grown food, almost all fruits and vegetables contain small amounts of pesticides. At low levels they are not really toxic or dangerous. But indeed the levels must be low and they must be controlled. Now suppose our purpose is determination of pesticide content in this orange and now come the questions. Oranges are conserved with pesticides on the surface. So in principle inside orange, there should be no pesticides at all. On the other hand, pesticides can diffuse from the orange peel to the inside. Therefore, the first question that comes is: whether we just determine pesticides on the orange surface or also in the inside ? Maybe we should determine pesticides in the overall orange. But let's look what orange looks like from the inside. We see that it's a far from homogeneous material. At the outside we have the peel and you see even the peel itself has two layers: one is yellowish the other is whitish. Then comes the orange fruit part and finally the inside there's again a white matter and also between these fruit slices there are some other other kinds of material. Meaning this orange has numerous different components, actually numerous different materials involved, and the pesticide content in each of them can be fully different. Therefore, for defining the measurand in the case of analysis of pesticides in the orange, the following decisions have to be made: whether its overall orange or some part of it ? If it is some part, then which part ? If it's only peel, then which layer of the peel ? So we see, there are important questions and all these need to be addressed and clearly answered, if we want to get our measurand right. Now, this is even not yet enough, let us think. We may be interested in our measurand in this one single particular orange. But on the other hand we may also be interested in the whole lot of oranges where this particular orange or perhaps few oranges have been taken from a sample. Therefore we also must ask the question: whether our measurement result relates only to the sample that we took from the whole body of the material or whether it refers to the whole body of material ? And now any of these different parts or the overall orange can be combined with this either sample or full, so that we can in principle have several measurand definitions just for this analytical task of determining some pesticide content in the orange. And let us look how they could be classified. Only sample. Full lot. So we see that we now have a matrix of eight different possibilities. And the measurand definition must clearly say into which of these eight slots we aim to get. |
Nous allons maintenant regarder certains principes de bases pour l'estimation d'incertitudes. Le principal d'entre eux est la définition de mesurande. C'est un concept très important qui est parfois un peu négligé. Je vais expliquer pourquoi il est extrêmement important de vraiment définir clairement ce qu'est le mesurande et je vais le faire à l'aide d'un exemple. Prenons cette orange, et supposons que notre objectif, notre but, soit de déterminer si cette orange est propre à la consommation, en déterminant sa teneur en pesticides. Cela implique de savoir si la concentration en pesticides est suffisamment faible dans cette orange. De nos jours, à moins qu'ils soient issus de l'agriculture biologique, presque tous les fruits et légumes contiennent une petite quantité de pesticides. En faibles quantités, ces derniers ne sont pas vraiment nocifs. Mais leurs concentrations doivent être suffisamment basses et elles doivent être surveillées (dans le temps). Maintenant, supposons que notre objectif soit la détermination de la teneur en pesticides contenue dans cette orange, alors des questions se posent. Les oranges contiennent des pesticides à leur surface. Donc, en principe, à l'intérieur de l'orange, il ne devrait pas y avoir de pesticides du tout. Cependant, les pesticides peuvent diffuser de la peau de l'orange jusqu'à l'intérieur de celle-ci. Donc, la première question qui se pose est : faut-il juste déterminer la quantité de pesticides à la surface de l'orange ou bien aussi à l'intérieur ? Peut-être faut-il déterminer la quantité de pesticides dans toute l'orange (peau + intérieur) ? Regardons à quoi une orange ressemble à l'intérieur. Nous voyons que cette dernière est loin d'être homogène. A l'extérieur, nous avons la peau et vous voyez que la peau est elle-même constituée de deux couches : l'une est jaunâtre et l'autre blanchâtre. Ensuite, nous avons le fruit de l'orange. Au centre, il y a finalement de nouveau une matière blanche, et il y a également, entre les tranches de fruits, une autre sorte de matière. Ce qui veut dire que cette orange contient de nombreux composants différents. En fait, de nombreuses parties différentes sont impliquées et la teneur en pesticides dans chacune d'elles peut-être très différente. Donc, pour définir le mesurande, pour cette analyse de pesticides dans une orange, la décision à la question suivante doit être prise : analyse-t-on l'orange en entier ou bien une partie de cette dernière ? Et si c'est une partie, laquelle ? Est-ce uniquement la peau, et si oui, quelle couche de la peau ? Nous voyons bien qu'il y a des questions importantes et qui nécessite d'y trouver une réponse claire si on souhaite définir comme il le faut notre mesurande. Cependant, ce n'est pas encore suffisant. Réfléchissons. Pour notre mesurande, on peut vouloir s'intéresser à cette unique orange. Mais on peut également vouloir s'intéresser au lot entier d'orange dans lequel celle-ci où seulement quelques oranges ont été prélevées. Donc, nous devons aussi nous poser la question : notre résultat de mesure se rapporte-t-il uniquement à l'échantillon prélevé sur l'ensemble de la population ou se réfère-t-il à l'ensemble de la population ? Maintenant, chaque constituant (de l'orange), ainsi que l'orange elle-même, peuvent être combinés avec soit l'échantillon seul ou tout le lot, ce qui peut, en principe, donner plusieurs définitions du mesurande, rien que pour cette tâche analytique qui est la détermination de la teneur en pesticides dans une orange. Et regardons comment ces mesurandes peuvent être classifiés. Echantillon seul ? Lot entier ? Nous avons maintenant un tableau avec huit mesurandes différents possibles. Le mesurande que l'on aura défini doit indiquer très clairement à laquelle de ces huit situations, on souhaite apporter une réponse. |
Définir le mesurande dans le cas de la détermination de la teneur en pesticides dans les oranges n’est pas trivial. D'une part, il est important de définir si le résultat est appliqué à une seule orange ou à quelques oranges prises comme échantillon ou s'il s'applique à l'ensemble du lot d'oranges (l'objet d'analyse dans son ensemble, également appelé cible d'échantillonnage). D'autre part, les oranges ne sont pas homogènes.
Les pesticides sont appliqués sur une surface d'orange, pas à l'intérieur. Ainsi, les pesticides peuvent diffuser de la peau d'orange vers l'intérieur. Il existe donc différentes possibilités: orange entière, peau entière, partie extérieure de la peau, chair d'orange uniquement.
En combinant ensemble, nous avons 8 possibilités, dans lesquelles nous pouvons déterminer exactement les pesticides. La mesure de la teneur en pesticides selon chacune des possibilités conduira à des résultats différents et non comparables entre eux.
En outre (non expliqué dans la vidéo), au lieu de définir le mesurande via le contenu total en analyte de l'échantillon (ou d'une partie de l'échantillon), il est souvent plus pratique d'examiner uniquement une partie de l'analyte.
Un bon exemple est la détermination du phosphore dans le sol. Bien qu'il soit possible de déterminer la teneur en phosphore totale dans le sol, il est en fait plus intéressant de déterminer uniquement la partie disponible pour les plantes - le phosphore biodisponible - car c'est cette partie de la teneur en phosphore totale qui contribue à la fertilité du sol et présente donc un intérêt pour l’agriculture.
La teneur en phosphore totale et la teneur en phosphore biodisponible sont des quantités différentes et leurs valeurs pour le même sol diffèrent fortement. Cela a des implications importantes pour la procédure de mesure. Ceci sera expliqué dans le 5.2.
6.2. Procédure de mesure
https://sisu.ut.ee/measurement/52-measurement-procedure
Résumé : Les principales étapes d’une procédure de mesure / analyse sont présentées sur l’exemple de la mesure d’un pesticide: Préparation de l’échantillon (dans ce cas: homogénéisation, extraction (s), purification des extraits), étalonnage de l’instrument, analyse réelle. Il est souligné que la procédure de mesure doit correspondre à la définition du mesurande.
Procédure de mesure
http://www.uttv.ee/naita?id=17586
| The second principle of mesurement uncertainty estimation is about measurment procedure or analysis procedure. The essence of this principle is, that the measurment procedure must correspond to the measurand definition. So if we determine the pestcide content only in the peel, then obviously we have to separate this peel from the reste of the orange. If on the other hand we want to determine the pesticide content in the overall orange we must analyse the overall orange. And since most probably pesticide content is different in different part of the orange, we must homogenize it carefuly. So the measurement procedure must make every effort to realy correspond to the measurand definition. Let us briefly look what the measurement procedure look like if we want determine pesticide content in orange. And suppose we want to determine it in the overall orange meaning or parts of it include. The first part of this procedure almost always is homogenization. This is usually followed by some kind of extraction or even several extractions. Into those extractions, care is taken as to extract the pesticide of interest into our sample usualy and lead all the other components of orange into the waste. Obviously orange is a very complexe matrix meaning it as a lot of components, and the most of those for us are only nuisance They are not neacessary. We want to get only the analyte out of that orange. Depending on how the procedure goes, the next part of it can be purification of the extract. And sometimes also concentration. And if that is done, then with this extract that we finally have, which contains as much as possible of the pesticides that were in the orange, and as little as possible of the others compounds. Is then analysed with the suitable analitical technique. Each and every of this steps introduces some uncertainty and all those uncertainty sources that are here in those steps have to be carefuly considered and those that their are important need to be taken into acount. And this will we look in the separate lecture. | Le second principe de l'estimation de l'incertitude de mesure traite de la procédure de mesure ou de la procédure d'analyse. L'essence même de ce principe est que la procédure de mesure doit correspondre à la définition de mesurande. Donc si nous déterminons la teneur en pesticide seulement dans la peau d’une orange, alors nous devons évidemment séparer cette peau du reste de l'orange. Si par contre nous voulons déterminer la teneur en pesticide de l'orange dans sa globalité, nous devons analyser l'entièreté de celle-ci. Et puisque la teneur en pesticide est très probablement différente dans les différentes parties de l'orange, nous devons l'homogénéiser avec soin. La procédure de mesure doit donc s'efforcer de correspondre réellement à la définition du mesurande. Regardons brièvement à quoi la procédure de mesure ressemble, si nous voulons déterminer la teneur en pesticide dans l'orange, et supposons que nous voulions la déterminer dans l'orange entière ou dans un morceau de celle-ci. La première partie de cette procédure est presque toujours l'homogénéisation. Elle est souvent suivie par une sorte d'extraction ou même plusieurs extractions. Dans ces extractions, nous prenons soin d'extraire le pesticide qui nous intéresse dans notre échantillon et de diriger tous les autres composants de l'orange dans les déchets. Il est évident que l'orange est une matrice très complexe, ce qui signifie qu'elle possède beaucoup de composants, et la plupart de ceux-ci ne sont pour nous que des nuisances. Ils ne sont pas nécessaires. Nous voulons extraire uniquement l'analyte de l'orange. En fonction du déroulement de la procédure, la prochaine partie peut être la purification de l'extrait, et parfois aussi la concentration (de l'extrait). Et si cela est fini, alors cet extrait que nous avons finalement obtenu, contient le plus possible de pesticides qui étaient dans l'orange, et le moins possible des autres composés. Il est alors analysé avec la technique analytique souhaitée. Chacune de ces étapes introduisent une certaine incertitude et toutes ces sources d'incertitude doivent être considérées avec soin et celles qui sont importantes doivent être prises en compte. Et c'est ce que nous verrons dans une autre conférence. |
Ce schéma d'une procédure d'analyse chimique est très général. Dans des cas spécifiques, il peut y avoir des écarts par rapport à ce schéma (plus ou moins d'étapes). En particulier, l’échantillonnage n’est pas présenté ici comme une étape de la procédure d’analyse chimique. Ceci est valable si les échantillons sont amenés au laboratoire pour analyse et que le laboratoire lui-même ne procède pas à l'échantillonnage (voir le chapitre 5.3 pour plus de détails).
Il convient de souligner l’importance de la préparation des échantillons en tant qu’étape de la procédure analytique. La majorité des procédures analytiques exigent que l’échantillon soit converti en une solution contenant la plus grande part possible de l’analyte issu de l’échantillon (idéalement la totalité) et le moins possible d’autres composants de la matrice de l’échantillon. En chimie analytique, la matrice d'échantillon est le terme utilisé pour décrire conjointement tous les composants de l'échantillon, à l'exception du ou des analytes. Les composants de la matrice agissent souvent comme des composés interférents, ce qui peut augmenter ou diminuer artificiellement le résultat. Par conséquent, il est important de minimiser leur contenu dans la solution obtenue à partir de l'échantillon. Si les composés interférents ne peuvent pas être totalement éliminés et que les interférences ne peuvent pas être corrigées (ce qui est tout à fait habituel dans l'analyse chimique), alors leur effet doit être pris en compte dans l'estimation de l'incertitude de mesure.
La préparation des échantillons est souvent la partie de l'analyse chimique qui demande le plus de travail et, dans la plupart des cas, c'est également la partie qui présente la plus grande contribution à l'incertitude. La préparation des échantillons implique généralement l'une des deux approches suivantes:
- Détruire essentiellement la matrice d'échantillon de manière à obtenir une solution contenant le ou les analyte(s) et quelques composants de la matrice. Cela se fait souvent par digestion avec des acides ou par fusion avec des alcalins ou des sels. Cette approche convient à la détermination d'éléments.
- Séparer le ou les analyte(s) de la matrice d'échantillon afin d'obtenir une solution contenant le ou les analyte(s) où la quantité de composants de la matrice est aussi faible que possible. Cela se fait généralement par un ensemble d'extractions. Cette approche convient aux analytes organiques.
Évidemment, le choix de la procédure de préparation de l’échantillon dépend du fait que le mesurande correspond au contenu total en analyte de l’échantillon ou à une partie de celui-ci, par exemple la teneur en analyte biodisponible (voir le texte du chapitre 5.1). Dans le cas de, par exemple la détermination de la teneur en phosphore totale dans le sol, l'analyste peut choisir la procédure de préparation de l'échantillon. Toutes les procédures menant à la détermination de la teneur en phosphore totale (impliquant souvent la destruction complète de la matrice) conviennent. Dans le cas de la détermination de la teneur en phosphore biodisponible dans le sol, la procédure de préparation de l'échantillon doit imiter la façon dont les plantes absorbent le phosphore du sol. La préparation des échantillons implique donc un lessivage dans des conditions prédéfinies. Ces procédures de préparation des échantillons sont souvent normalisées et chaque fois que les résultats sont censés être comparables, ils doivent être obtenus avec la même procédure. Ainsi, dans ce dernier cas, la procédure de préparation de l’échantillon devient partie intégrante de la définition du mesurande.
6.3. Sources d'incertitude de mesure
https://sisu.ut.ee/measurement/53-sources-uncertainty
Résumé : L'aperçu des sources d'incertitude possibles est présenté via l'exemple de l'analyse de pesticide. Bien que les sources d'incertitude soient présentées via l'exemple d'analyse des pesticides, les mêmes sources d'incertitude sont valables pour la majorité des autres méthodes d'analyse. La plupart des sources d'incertitude sont liées à des étapes spécifiques de la procédure d'analyse. Il est souligné que la préparation des échantillons est généralement le facteur qui contribue le plus à l’incertitude de mesure. Lors de la réalisation d'une analyse chimique, il convient de veiller à minimiser (de préférence éliminer) l'influence des sources d'incertitude, dans la mesure du possible. Et ce qui ne peut être éliminé doit être pris en compte. Il n'est pas nécessaire de quantifier chaque source d'incertitude individuellement. Au lieu de cela, il est souvent plus pratique de quantifier conjointement plusieurs sources d’incertitude.
Sources d'incertitude de mesure
http://www.uttv.ee/naita?id=17587
| The next principle of measurement uncertainty estimation is that all relevant uncertainty sources have to be considered and those that are important have to be taken into account. We have already touched the concept of uncertainty source, but let us now go a little bit deeper into it, and let us do that again on the example of this orange. Pesticides determination of orange. And let us again look at the steps that are in chemical analysis, and let us try to think what types of uncertainty, or what kinds of uncertainty sources can be involved in different steps. And let us now go back to the place where the sample was taken, and we start with the first operation that is inherent in chemical analysis and this is sampling. When sample is taken, then the sample is brought to the laboratory, and depending on its nature, it is usually reduced in size because initial samples can be very large. And then, different operations are done, and as we've already seen in the case of orange we then usually need to homogenize, then usually some extraction, then purification and concentration of the extract. And then when we finally have the extract solution that can be introduced into the analytical device, into the analytical instrument, we really can carry out the instrumental analysis. There's one more important step that is not evident here now, most, the large majority of those analytical instruments need calibration, calibration with the analyte and this also needs to be done. Finally, we do data treatment and get the result. This could be, in a nutshell, the main operations that are involved in this pesticide determination in orange. It's important to note that chemical analysis is a very diverse range of activities. So that not always these same steps apply, some can be omitted and some new steps can be there, but as an example it pretty well summarizes the general situation in chemical analysis. Let us now look at the potential uncertainty sources that are involved in these steps. If we start from sampling, then sampling does have uncertainty due to non representativeness on the sample. Meaning the pesticide content in the particular oranges that we took from the overall lot can be somewhat different than the average concentration in the other oranges in that lot, and in order to avoid that, people usually take samples in such a way that they take sample from many parts of the whole lot and then average in the laboratory. This is for assuring that the sample is as representative as possible. It is also important to note in the case of sampling, that this uncertainty source only will be taken into account if we present our result for the whole bulk, the whole lot of the analysis object. If we present our result only for the sampling, then the uncertainty source due to non representativeness of the sample is not taken into account. Let us now look at the next steps. These next steps can jointly be called "sample preparation". And sample preparation has numerous uncertainty sources. First of all, the analyte can, in different ways, be lost so that during all these operations, some part of the analyte is lost and finally less analyte arrives into the instrument than was actually present in the sample; and those losses can be due to the following : incomplete extraction, losses during purification or concentration, some analytes are prone to decomposition. These losses during purification can also be for example caused by adsorption on surfaces. In some cases analytes can be volatile and then there can be volatilization, pesticides are almost never volatile but otherwise it can be. Now, all these effects that I have mentioned here lead to decrease the measurement result, but there are effects that also can lead to increase of the analysis results. And in this particular section, this can be caused by contamination. It is generally accepted and recognized that sample preparation is usually the most important part of chemical analysis, from the point of view of a measurement uncertainty, meaning : there are more uncertainty sources and they are usually more influential in this sample preparation part than in any others except perhaps sampling, sampling uncertainty also can be very very high. Now let's look further in this instrumental analysis part, we have different instrument relative uncertainty sources such as instrument noise, drift, etc... But on the other hand, very importantly, in this part also there's the incomplete selectivity source. This uncertainty source means that instrument is also influenced by some other compound besides the analyte. As is generally known in analytical chemistry selectivity is one of the most important things, if we determine some concrete pesticides in orange, then we are interested that the signals that we get from the instrument really do correspond to these concrete pesticides but not to other compounds that perhaps were present in orange. But almost always those other compounds have some small influence on the signal, thereby causing uncertainty and very often those influences are positive, in the sense that they increase the signal so that we get the seemingly higher result. This kind of uncertainty source and also many of those that are listed here are especially problematic and especially serious if we determine low levels of analytes, and pesticide analysis certainly is a low level analysis or as sometimes people say : trace analysis. Now, calibration. It is first of all influenced by all instrumental sources; and on the other hand, it is also influenced by the concentration and purity of the calibration materials. And data treatment can also have its uncertainties. It can sometimes be not so easy to extract the concrete information, the concrete signal from the data that is obtained by the instrument, so certain data treatment uncertainties are also present, such as for example integration of chromatographic pics. If you look now at this list, something is missing. And we see that completely missing are the uncertainty sources due to pipettes, volumetric flasks, weighing, etc... But they also of course need to be considered. The reason why they are not directly introduced here is and they occur in many places, so whenever sample preparation is done, it usually involves some weighing after sample size is reduced. It's weighed and weighing uncertainty is there. Throughout these operations, some solutions are used and oftentimes volumes are measured, and of course calibration solutions are made usually by weighing and volumetric operations. So that both weighing and volumetric uncertainty sources, we can also put here as general uncertainty sources that influence several parts here. Now what should an analytical chemist do with this uncertainty sources? There are two main things that we have to consider and we have to do with them. First of all, and very importantly, this whole procedure, and the way it is executed, has to be done in such a way as to minimize all these uncertainty sources. It is good if they can be eliminated altogether, but if they cannot be eliminated, making them as small as possible is also useful, and on the other hand, every care must be taken to find out which of them are important uncertainty sources, and those that are important have to be quantified. And again care has to be taken that they are quantified in a rigorous and logical manner. | Le prochain principe de l'estimation de l'incertitude de mesure est le suivant : toutes les sources d'incertitudes pertinentes doivent être considérées et celles qui sont importantes doivent être prises en compte. Nous avons déjà abordé le concept de source d'incertitude mais nous allons maintenant l'approfondir un peu plus grâce, à nouveau, à l'exemple de cette orange. La détermination de pesticides de l'orange. Regardons encore une fois les étapes d'analyse chimique et essayons de réfléchir à quels types d'incertitudes, ou quelles sources d'incertitudes peuvent être impliquées dans les différentes étapes. Revenons maintenant à l'endroit où l'échantillon a été prélevé, nous commençons avec la première opération inhérente à l'analyse chimique : l'échantillonnage. Quand l'échantillon est prélevé, il est apporté au laboratoire, et selon sa nature, il est souvent réduit en taille car les échantillons initiaux peuvent être très grands. Puis différentes opérations sont effectuées, et comme nous l'avons déjà vu dans le cas de l'orange nous devons généralement homogénéiser, puis habituellement procéder à une ou plusieurs extraction(s), à une purification et enfin à une concentration de l'extrait. Et ensuite quand nous avons enfin la solution extraite qui peut être introduite dans l'appareil d'analyse, dans l'instrument analytique, nous pouvons réellement réaliser l'analyse instrumentale. Il y a une autre étape importante qui n'est pas évidente, ici, la majorité de ces instruments analytiques ont besoin d'être calibrés avec un analyte et cela doit également être fait. Finalement nous faisons du traitement de données et obtenons le résultat. Cela pourrait être, en résumé, les principales opérations qui sont impliquées dans cette détermination des pesticides dans l'orange, et il est important de noter que l'analyse chimique a une gamme d'activités très diversifiée. Ces mêmes étapes ne s'appliquent pas toujours, certaines peuvent être omises et d'autres nouvelles étapes peuvent être ajoutées, mais cet exemple résume plutôt bien le cas général de l'analyse chimique. Etudions maintenant les sources potentielles d'incertitude qui interviennent dans ces étapes. Si nous commençons par l'échantillonnage, celui-ci possède une incertitude due à la non-représentativité de l'échantillon. Ce qui signifie que la teneur en pesticides des oranges particulières que nous avons prises dans le lot complet peut être légèrement différente de la concentration moyenne dans les autres oranges de ce lot, et pour éviter cette situation, on prélève généralement des échantillons à différents endroits du lot entier (d'oranges) et on fait ensuite la moyenne au laboratoire pour s'assurer que l'échantillon est le plus représentatif possible. Il est aussi important de noter que dans le cas de l'échantillonnage, que cette source d'incertitude ne sera prise en compte que si nous présentons nos résultats pour l'ensemble du lot, pour le lot entier d'objets d'analyse, et si nous présentons nos résultats seulement pour l'échantillonnage, alors la source d'incertitude due à la non-représentativité de l'échantillon n'est pas prise en compte. Passons maintenant aux étapes suivantes. Les prochaines étapes peuvent être conjointement appelées "la préparation de l'échantillon". Et la préparation de l'échantillon possède de nombreuses sources d'incertitude. Premièrement, l'analyte peut être perdu de différentes façons, de telle sorte que pendant toutes les opérations une partie de l'analyte est perdue et que finalement une quantité moins importante d'analyte arrive dans l'instrument comparé à ce qui était réellement présent dans l'échantillon, et ces pertes peuvent être dues aux éléments suivants : à une extraction incomplète, à des pertes durant la purification ou la concentration, certains analytes sont susceptibles de se décomposer. Ces pertes lors de la purification peuvent également être causées par exemple par l'adsorption sur les surfaces. Dans certains cas les analytes peuvent être volatils et il peut ensuite y avoir la volatilisation (les pesticides ne sont presque jamais volatils mais d'autres analytes peuvent l'être). Tous ces effets que j'ai mentionné ici entraînent une diminution des résultats de mesures, mais certains effets peuvent aussi mener à une augmentation des résultats d'analyse. Et dans cette section particulière, cela peut être causé par la contamination. Il est généralement admis et reconnu que la préparation de l'échantillon est souvent la partie la plus importante de l'analyse chimique du point de vue de l'incertitude de mesure : il y a plus de sources d'incertitude et elles sont généralement plus influentes dans cette partie de la préparation de l'échantillon que dans toutes les autres sauf peut-être lors de l'échantillonnage, où l'incertitude de l'échantillonnage peut également être très très élevée. Maintenant regardons plus en détails cette partie de l'analyse instrumentale : nous avons différentes sources d'incertitude relative aux instruments comme le bruit, la dérive, etc… Mais d'un autre côté, la source de sélectivité incomplète est également très importante dans cette partie. Cette source d'incertitude signifie que l'instrument est aussi influencé par un autre composé que l'analyte. Comme il est généralement connu en chimie analytique, la sélectivité est l'une des choses les plus importantes. Si nous déterminons la concentration de certains pesticides concrets dans l'orange, nous souhaitons que les signaux que nous recevons de l'instrument correspondent réellement aux signaux de ces pesticides concrets et non à d'autres composés qui étaient peut-être présents dans l'orange. Mais dans de nombreux cas, ces autres composés ont une petite influence sur le signal ce qui entraîne une incertitude et, très souvent, ces influences sont positives, dans le sens où elles augmentent le signal de telle manière que nous obtenons un résultat qui semble plus élevé. Ce type de source d'incertitude et aussi beaucoup de celles qui sont citées ici sont particulièrement problématiques et très importantes si nous déterminons de faibles niveaux d'analytes. L'analyse des pesticides est certainement une analyse de faible niveau ou comme certains la qualifient : c'est une analyse de traces. Maintenant, la calibration. Tout d'abord, la calibration est influencée par les sources instrumentales, et d'autre part, elle est influencée par la concentration et la pureté des matériaux de calibration. Le traitement des données peut également avoir ces incertitudes. Il est parfois difficile d'extraire une information concrète, comme le signal des données qui sont obtenues par l'instrument. Donc certaines incertitudes de traitement des données sont aussi présentes, comme par exemple lors de l'intégration de pics chromatographiques. Si vous regardez cette liste vous constaterez qu'il manque quelque chose : les sources d'incertitude dues aux pipettes, aux fioles jaugées, à la pesée etc… Mais, il faut aussi, bien sûr, les considérer. La raison pour laquelle elles ne sont pas directement introduites est qu'elles ont lieu dans de nombreuses étapes. Dès que la préparation de l'échantillon est faite, elle fait intervenir une pesée après la réduction de la taille de l'échantillon, une incertitude de pesée est donc présente. Au cours de ces opérations, certaines solutions sont utilisées, souvent des volumes sont mesurés, et bien sur, les solutions de calibration sont généralement réalisées par des opérations de pesée et/ou de volumétrie ce qui fait que les sources d'incertitude de pesée et de volumétrie peuvent être considérées comme des sources d'incertitude générales qui influencent plusieurs étapes. Maintenant, que doit faire un analyste avec ces sources d'incertitude? Il y a deux choses principales que nous devons considérer et faire avec. En premier lieu et c'est très important, toute cette procédure et la façon dont elle est exécutée doit être faite de manière à minimiser toutes ces sources d'incertitude. C'est bien si elles peuvent être toutes éliminées, mais si on ne peut pas, il faut les rendre les plus faibles possibles, et d'autre part, il faut tout mettre en œuvre pour déterminer lesquelles sont des sources d'incertitude importantes et celles qui le sont doivent être quantifiées. Et là encore, il faut veiller à ce qu'elles soient quantifiées de façon rigoureuse et logique. |
Autoévaluation sur cette partie du cours : Test 5.3
6.4. Traitement des effets aléatoires et systématiques
https://sisu.ut.ee/measurement/54-treatment-random-and-systematic-effects
Résumé : Bien que, dans une série de mesures, les effets aléatoires et systématiques influent différemment sur les résultats des mesures, ils sont mathématiquement pris en compte de la même manière - en tant que composants d’incertitude présentés sous forme d’incertitudes types.
Traitement des effets aléatoires et systématiques
http://www.uttv.ee/naita?id=17712
| The next principle is about treatment of random and systemic effects and the principle says that random and systemic effects are treated the same way. It is principle number four. And let us look at this principle on the example of piped things that we did one of the in one of the previous lectures. Here we had different uncertainty sources namely three different uncertainty sources: uncertainty due to repeatability, uncertainty due to calibration of the pipette and uncertainty due to temperature. Let us think which of these are related to random effects and which are related to systemic effects. So obviously repeatability is a random effect by definition because it takes into account the small differences that occur from pipetting to piped. So this is random. Now this calibration uncertainty. If we use the same pipette for the different pipetting operations is a typical systematic uncertainty source because if the mark is a little bit off from 10 milliliters then it remains off for all those pipetting operations. So that calibration effect is a typical systemic effect. Now temperature is more tricky than any of these two. If the temperature during the day remains constant then it is a typical systemic effect however if the temperature changes from measurement to measurement then it actually becomes a random effect. So the temperature can be either a random or a systematic effect. And in spite of these effects being different by their image they are still all accounted for exactly the same way all these contributions are estimated converted into standard uncertainties and standard uncertainties are then combined. It is important to stress that those concepts random and systematic are in fact not absolute concept. An effect that under one set of conditions is systematic can under different set of conditions be a random effect. For example, as long as we do pipetting using the same pipette, this calibration effect remains a systemic effect but if for every pipetting operation we take a new pipette then in fact the calibration uncertainty becomes a random effect because in some cases the mark is slightly below in some cases slightly above 10 milliliters. And how systemic effects can be converted into random effects by changing conditions will be explained in a coming lecture. | Le prochain principe porte sur le traitement d'effets aléatoires et systématiques. Le principe cite que les effets aléatoires et systématiques sont traités de la même manière. C'est le principe numéro quatre. Regardons ce principe sur l'exemple de choses pipetées comme nous avions fait dans une des lectures précédentes. Là nous avions différentes sources d'incertitudes, notamment, trois différentes sources d'incertitudes: une incertitude due à la répétabilité, une incertitude due à la calibration de la pipette et une incertitude due à la température. Réfléchissons à lesquelles de celle-ci sont liées à des effets aléatoires et lesquelles sont liées à des effets systématiques. Alors, évidemment, la répétabilité est un effet aléatoire par définition car elle prend en compte les petites différences qui apparaissent à chaque pipettage. Donc c'est bien aléatoire. Maintenant pour l'incertitude de calibration. Si nous utilisons la même pipette pour les différentes opérations de pipettage, il s'agit d'une source d'incertitude systématique typique car si la marque est un peu décalée, de 10 mL, alors elle reste décalée pour toutes ces opérations de pipettage. Donc cet effet de calibration est un effet systématique typique. Pour la température c'est plus compliqué que les deux précédents. Si la température reste constant au cours de la journée, c'est alors un effet systématique typique. Cependant, si la température évolue d'une mesure à l'autre alors cela devient en fait un effet aléatoire. Donc la température peut être soit un effet systématique ou aléatoire. Et malgré que ces effets soient différents, ils sont encore tous comptabilisés de la même manière. Toutes ces contributions sont estimées et converties en écart-types et les écart-types sont alors combinés. Il est important d'insister sur le fait que ces concepts, aléatoires et systématiques, sont en fait, pas des concepts absolus. Un effet, qui sous certaines conditions est systématique, peut, sous d'autres conditions, être un effet aléatoire. Par exemple, tant que nous pippettons en utilisant la même pipette, cette effet de calibration reste un effet systématique, mais si pour chaque opération de pipettage nous prenons une nouvelle pipette, alors l'incertitude de calibration devient un effet aléatoire car la marque est légèrement en dessous 10 mL dans certain cas et légèrement au dessus dans d'autres. Et de quelles manières ces effets systématiques peuvent être convertis en effets aléatoires en changeant des conditions va être expliqué dans une prochaine lecture. |
Dans le cas du pipetage (démontré et expliqué aux chapitres 2 et 4.1), il existe trois sources principales d’incertitude: la répétabilité, l’incertitude de calibration de la pipette et l’effet de la température. Ces effets influencent le pipetage de différentes manières.
- La répétabilité est un effet aléatoire typique. Chaque opération de pipetage est influencée par des effets aléatoires qui entraînent les différences entre les volumes pipettés dans des conditions identiques.
- L'incertitude liée à l'étalonnage de la pipette est un effet systématique typique: si au lieu de 10,00 ml, le repère sur la pipette est, par exemple, à 10,01 ml, le volume de la pipette sera systématiquement trop élevé. Cela signifie que, même si les résultats de pipetage individuels peuvent être inférieurs à 10,01 ml (et même inférieurs à 10,00 ml), le volume moyen sera supérieur à 10,00 ml: environ 10,01 ml.
- L'effet de la température peut être, selon la situation, soit systématique, soit aléatoire, soit (très souvent) un mélange des deux. Cela dépend de la stabilité de la température pendant les répétitions (qui est influencée par la durée totale de l'expérience).
Bien que les trois sources d'incertitude influencent les résultats de pipetage de différentes manières, elles sont toutes prises en compte de la même manière, via des contributions d'incertitude exprimées sous forme d'incertitudes type.
En principe, il est possible d'étudier les effets systématiques, de déterminer leur ampleur et de les prendre en compte en corrigeant les résultats. Lorsque cela est réalisable, cela devrait être fait. Si cela n’est pas fait, les résultats seront biaisés, c’est-à-dire qu’ils seront systématiquement décalés par rapport à la valeur vraie.
Un exemple dans lequel l'effet systématique peut être déterminé et la correction introduite avec un effort raisonnable est l'étalonnage de la pipette, expliqué dans l'exemple du chapitre 4. Deux cas ont été examinés: sans correction et avec correction:
- Dans le chapitre 4.1, l’incertitude d’étalonnage de ± 0,03 ml spécifiée par le producteur est utilisée. Ceci correspond à la situation dans laquelle il existe éventuellement un effet systématique. La différence [1] possible du volume réel de la pipette par rapport à son volume nominal elle n’est ni examinée ni corrigée de manière approfondie et l’incertitude de ± 0,03 ml lui est attribuée. laquelle à une très forte probabilité de prendre en compte cet effet. En conséquence, l’incertitude type de l’étalonnage était u (V, CAL) = 0,017 ml (une distribution rectangulaire est supposée).
- Le chapitre 4.6 montre comment déterminer le volume réel de la pipette par calibrage. Il a été trouvé que le volume de la pipette était de 10,006 ml. L’étalonnage effectué en laboratoire comporte encore des incertitudes, mais cette incertitude est maintenant due à la répétabilité pendant l’étalonnage (effets aléatoires) et est presque 10 fois plus petite: u (V, CAL) = 0,0018 ml.
Ainsi, lorsque la correction des effets systématiques peut être effectuée avec un effort raisonnable, elle peut entraîner une diminution significative de l’incertitude de mesure. Cependant, dans de nombreux cas, la détermination exacte d'un effet systématique (détermination exacte du biais) peut impliquer un effort très important et, de ce fait, peut s'avérer peu pratique. Il peut également arriver que l’incertitude de correction ne soit pas beaucoup plus petite que l’incertitude due à un biais possible.
Autoévaluation sur cette partie du cours : Test 5.4
https://sisu.ut.ee/measurement/node/2283
***
[1] Le mot possible signifie ici qu’en fait, il peut ne pas y avoir d’effet systématique - le volume réel de la pipette peut être de 10,00 ml. Nous ne le savons tout simplement pas.
7. Retour sur les effets aléatoires et systématiques
https://sisu.ut.ee/measurement/6-random-and-systematic-effects-revisited
Résumé : Ce chapitre explique que l’impact d’un effet sur les résultats de la mesure, de manière aléatoire ou systématique, dépend des conditions. Les effets systématiques à court terme peuvent devenir aléatoires à long terme. C'est la raison pour laquelle la répétabilité est, par sa valeur, inférieure à la reproductibilité intra-laboratoire, et que celle-ci est à son tour inférieure à l'incertitude-type composée. Cette section explique également que les estimations de l’incertitude des types A et B ne correspondent pas un à un aux effets aléatoires et systématiques.
Comment un effet intra-journalier systématique peut-il devenir un effet aléatoire à long terme ?
http://www.uttv.ee/naita?id=17713
| In one of the earlier lectures, I mentioned that the concept of random and systematic effects are not absolute concept and that, under certain conditions, systematic effects can become random effects, this is something we will now look at more carefully. Here is a scheme of a measurement result. We have here concentration axes and, on day one, we have obtained these results. We have made five repeated measurements on that day, and their values are here. And now, these values are affected both by random and by systematic effects, and we can see that the random effect causes the differences between these values. They do not agree among themselves. But the systematic effect we don't see from here, because all those values are shifted from each other. And now, there are a number of systematic effects that, from day to day, can be different, and this is exactly the case here. So let us look what happens if we now do the same measurement on more days. See on day 2, all measurements were shifted from the result of the 1, and on day 5, they were off by this much. On day 9, they are off to here etc ... So we see that within each of those numerous days, that are depicted here, the systematic effects has different direction and different magnitude, and this means that, in the context of many days, in the context of long term, this effect is not any more a systematic effect but is a random effect, and if we now look at the histogram, that is formed from all these results, then it in fact represents quite nicely the normal distribution function , which means that the systematic effect also gives its contribution into the scatter of the results. and if we looked at the data in long term, we can treat this systematic effect as a random effect, and we can determine its magnitude by repeating measurements on different dates. And this, is as we will see in the coming lectures, is a highly useful possibility for practical measurement uncertainty estimation. This example is also useful for distinguishing between repeatability and we didn't have long term reproducibility, or as it is also called intermediate precision. So the repeatability causes the scatter within things. So, for example, on this day, due to repeatability, the scatter is this wide. Or day number 9, the scatter is around this wide. On day number 17, with the scatter is like this on 29th. Day number 22, the scatter is this one. So, we see in all those cases, the scatter is roughly this much but if we look at all those results together, then the within lab long term reproducibility or intermediate precision characterizes the scatter over all those days. So that the scatter, if expressed as intermediate precision, is very much wider than the scatter expressed by repeatability, and it's easy to see why it is wider. Into repeatability, this systematic effect does not go in but, in the intermediate precision, this systematic effect is also included. Therefore, the standard deviation of repeatability is always smaller, and sometimes very much smaller than the standard deviation within that long-term reproducibility or, as it is called, intermediate precision. And here, we can visually characterize those standard deviations. Repeatability standard deviation amount roughly this width. And intermediate precision standard deviation this width or half of the width. | Dans l'une des précédentes vidéos, j'ai mentionné que les concepts d'effets aléatoires et systématiques ne sont pas des concepts absolus et que, sous certaines conditions, les effets systématiques peuvent devenir des effets aléatoires, qui est quelque chose que nous allons voir maintenant plus attentivement. Voici un graphique d'un résultat de mesure. Nous avons ici en abscisse l'axe des concentrations et, au jour 1, nous avons obtenu ces résultats. Nous avons effectué cinq répétitions de mesures ce jour là, et ces valeurs sont ici. Et maintenant, ces valeurs sont affectées par des effets aléatoires et systématiques, et nous pouvons voir que l'effet aléatoire provoque des différences entre ces valeurs. Il n'y a pas d'accord entre eux. Nous ne voyons pas l'effet systématique ici, car toutes ces valeurs sont décalés les unes des autres. Et maintenant, il y a un nombre d'effets systématiques qui, de jour en jour, peut être différent, et qui est exactement le cas ici. Regardons ce qu'il se passe si nous faisons maintenant la même mesure sur plus de jours. Regardez au second jour, toutes les mesures étaient décalées par rapport au résultat du premier jour, et le jour 5, elles étaient loin de ce point. Au jour 9, elles étaient encore plus loin etc... Nous voyons donc qu'avec chacun de ces nombreux jours, qui sont représentés ici, les effets systématiques ont une direction et une amplitude différente, et cela signifie que, dans un contexte de plusieurs jours, dans un contexte de long terme, cet effet n'est plus un effet systématique mais est un effet aléatoire, et si nous regardons maintenant l'histogramme, qui est crée avec tous ces résultats, alors il représente en fait assez bien la fonction de la distribution normale, qui signifie que l'effet systématique donne également sa contribution à la dispersion des résultats. Et si nous regardions les données sur le long terme, nous pouvons traiter cet effet systématique comme un effet aléatoire, et nous pouvons déterminer son amplitude en répétant des mesures à différentes dates. C'est, comme nous le verrons dans les vidéo à venir, une possibilité très utile pour l'estimation de l'incertitude de mesure. Cet exemple est aussi utile pour faire la distinction entre la répétabilité et la reproductibilité à long terme (nous n'en avions pas), aussi appelée fidélité intermédiaire. La répétabilité provoque donc la dispersion Donc par exemple, à ce jour, en raison de la répétabilité, la dispersion fait cette largeur. Ou alors le jour 9, la dispersion fait environ cette largeur. Le jour 17, la dispersion est équivalente à celle du jour 29. Le jour 22, la dispersion fait celle-ci. Donc, nous voyons que dans tous ces cas, la dispersion est équivalente mais si nous regardons tous ces résultats simultanément, alors la reproductibilité à long terme au sein du laboratoire ou la fidélité intermédiaire caractérise la dispersion sur tous ces jours. Donc la dispersion, si elle est exprimée par la fidélité intermédiaire, est beaucoup plus large que la dispersion exprimée par la répétabilité, et c'est facile de voir pourquoi la dispersion est plus large. Pour la répétabilité, l'effet systématique n'entre pas en compte mais, pour la fidélité intermédiaire, cet effet systématique est inclus. Par conséquent, l'écart-type de répétabilité est toujours plus petit, et parfois beaucoup plus petit que l'écart-type de reproductibilité à long terme ou, comme on l'appelle, la fidélité intermédiaire. Ici, nous pouvons caractériser visuellement ces écarts-types. L'écart-type de répétabilité correspond à peu près à cette largeur. L'écart-type de fidélité intermédiaire correspond à cette largeur ou à la moitié de cette largeur. |
Effets aléatoires et systématiques à court terme et à long terme
Un effet systématique sur une courte période (par exemple dans la journée) peut être aléatoire sur une plus longue période. Exemples:
- Si un certain nombre d'opérations de pipetage sont effectuées dans la journée en utilisant la même pipette, la différence entre le volume réel de la pipette et son volume nominal (c'est-à-dire l'incertitude d'étalonnage) sera un effet systématique. Si le pipetage est effectué à des jours différents et que la même pipette est utilisée, c'est également un effet systématique. Cependant, si le pipetage est effectué à des jours différents et que différentes pipettes sont utilisées, cet effet se transformera en effet aléatoire.
- Un instrument est étalonné quotidiennement avec des solutions d'étalonnage faites à partir de la même solution mère, qui est refaite tous les mois. Dans ce cas, la différence entre la concentration réelle de la solution mère et sa concentration nominale est un effet systématique en un jour et également en quelques semaines. Mais sur une période plus longue, disons une demi-année, [1] Il ne peut pas être strictement défini ce que signifie «à long terme». Une indication approximative pourrait être: un an, «plusieurs mois» (au moins 4-5) est minimum. Bien sûr, cela dépend aussi de la procédure. cet effet devient aléatoire, car un certain nombre de solutions mères différentes auront été utilisées pendant cette période.
Conclusions:
- Un effet systématique à court terme peut être aléatoire à long terme;
- Plus le délai est long, plus les effets peuvent passer de systématique à aléatoire.
Comme expliqué dans une vidéo précédente, si la mesure du même échantillon ou d'un échantillon identique est répétée dans des conditions identiques (généralement dans la même journée) en utilisant la même procédure, l'écart type des résultats obtenus est appelé écart type de répétabilité et noté sr. Si la mesure d'un échantillon identique ou identique est répétée en utilisant la même procédure mais dans des conditions modifiées selon lesquelles les changements sont ceux qui ont lieu en laboratoire dans des pratiques de travail normales, l'écart-type des résultats est appelé reproductibilité intra-laboratoire ou fidélité intermédiaire. et il est noté sRW. [2]
Les conclusions exprimées ci-dessus sont la raison pour laquelle sr est plus petit que sRW. Simplement, certains effets qui dans la journée sont systématiques et ne sont pas pris en compte par sr deviennent aléatoires sur une plus longue période et sRW les prend en compte. L'incertitude-type composée uc est à son tour supérieure à la fidélité intermédiaire, car elle doit prendre en compte tous les effets significatifs qui influencent le résultat, y compris ceux qui restent systématiques également à long terme. Cette relation importante entre ces trois quantités est visualisée dans le schéma 6.1.

Schéma 6.1. Relations entre la répétabilité, la reproductibilité intra-laboratoire et l'incertitude composée
Les effets aléatoires et systématiques ne peuvent pas être considérés comme étant en relation un à un avec l'estimation de l'incertitude de type A et B. Ce sont des choses catégoriquement différentes. Les effets se réfèrent aux relations causales intrinsèques, tandis que l'estimation de l'incertitude de type A et B se réfère plutôt aux approches utilisées pour quantifier l'incertitude. Le tableau 6 illustre cela davantage.

Ainsi, en fonction de la chronologie, tous les effets provoquant l'incertitude peuvent être regroupés comme illustré dans le schéma 6.2. À court terme, la plupart des effets agissent comme systématiques et les effets aléatoires peuvent être quantifiés via la répétabilité. À long terme, plus (généralement la plupart) des effets sont aléatoires et peuvent être quantifiés via la reproductibilité intra-laboratoire (fidélité intermédiaire).
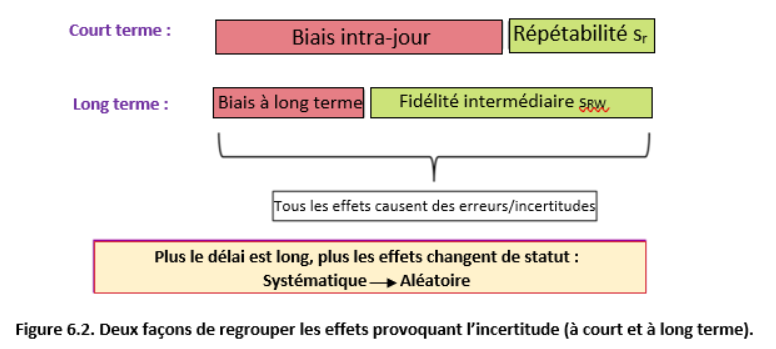
Comme expliqué dans le chapitre 8, les deux principales approches d'estimation de l'incertitude abordées dans ce cours utilisent les méthodes ci-dessus comme suit: L'approche de modélisation (ISO GUM) a tendance à suivre la vue à court terme (estimation de l'incertitude d'un résultat particulier sur un jour particulier), tandis que l'approche Single-lab (Nordtest) suit toujours la vision à long terme (estimation d'une incertitude moyenne de la procédure).
Déterminer la répétabilité et la reproductibilité intra-laboratoire en pratique
Les exigences types pour déterminer sr et sRW sont présentées dans le tableau 6.2.
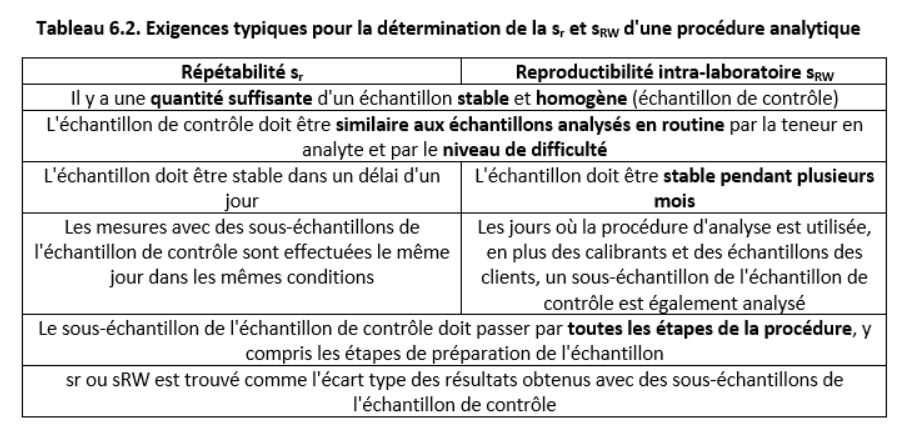
Lors de l'estimation des contributions d'incertitude dues aux effets aléatoires, il est important d'effectuer un certain nombre de mesures répétées. D'un autre coté, si par exemple la répétabilité de certaines procédures analytiques doit être estimée, chaque répétition doit couvrir toutes les étapes de la procédure, y compris la préparation des échantillons. Pour cette raison, faire de nombreuses répétitions demande beaucoup de travail. Dans cette situation, le concept d'écart type groupé devient très utile. Son essence est la mise en commun des écarts-types obtenus à partir d'un nombre limité de mesures. La vidéo suivante explique cela:
Ecart type groupé
http://www.uttv.ee/naita?id=18228
| As we've seen already, whenever we want to determine precision meaning repeatability or within lab long-term reproducibility, it is always important to make many parallel measurements. And in fact, the more we make, the better estimate of precision we get. Now oftentimes chemical analysis can be quite work intensive. Meaning it's not easy to make many parallel measurements especially not within a short time period. And here a statistical tool called pooled standard deviation becomes very helpful for us. So a pooled standard deviation can be used both for repeatability and within lab reproducibility calculation. And the main formula of pooled standard deviation calculation is here. So in the case of pooled standard deviation we always make measurements with several samples. And in this formula, these numbers here denote the sample numbers. So we have all together K samples. And each s is the standard deviation of that particular sample. S1 the standard deviation of the first, s2 of the second sample and so on. And n stand for the number of parallel measurements made with those samples. So all these standard deviations that we get with individual samples, we take to the square multiplied by the number of parallel measurements that we made minus one add all of them up and then divide by this kind of number, which is in fact the sum of all the parallel measurements numbers minus the number of samples. And what we eventually get is the pooled standard deviation. The good thing about pool standard deviation is that you can involve a large number of samples at the same time with each samples the number of measurements can be very small. In fact it is enough if with each sample just two measurements are made as long as you have many samples. And the number of degrees of freedom for pooled standard deviation is in fact this number here. So that if we have let's say ten samples but make with each of them only two measurements, then this number of degrees of freedom will be 10 times 2 minus 10 which is equal to 10. Meaning it's roughly statistically speaking as good as making 11 parallel measurements with the same sample. So, even though each of those standard deviations which are found from just two parallel measures is very very very very unreliable but if we have many of them and we average them according to this formula, then the resulting pooled standard deviation will be quite reliable in fact. It is important that all those samples that we carry out the measurements with, have to be similar. So that it has to be expected that their standard deviations are roughly comparable, roughly the same. If the samples have very different concentration levels, very different matrixes, very different level of difficulty of sample preparation, then the resulting pooled standard deviation will not carry much meaning. Now on the other hand, if we compare standard deviation that is obtained as a pooled standard deviation from a number of different samples, with in the case where with all the samples we make not so many parallel measurement, or the second case where we have just one single sample, and we make a large number of measurements with that samples, then in fact the pooled standard deviation can give us more reliable precision estimate because the properties of the slightly different samples will all be taken into account. So that the pooled standard deviation indeed will be an average standard deviation over all these samples. And secondly, usually those standard deviations are found on different days. Meaning on one day perhaps our instrument is in better working order on the other day it's in slightly worse working order. Again the resulting pooled standard deviation gives us the average situation in our lab. If we make standard deviation determination on just one day with just one sample, then it can one happen that this particular sample is a specifically good sample and the instrument on that they can be in a very good working condition which can result in a very low standard uncertainty which is a repeatability standard uncertainty in this case. And this can give us two optimistic estimate for the future use because whenever we determine such standard deviation we do this for using it in the future for some measurement uncertainty calculation. In a contrast pooled standard deviation gives us the average situation in our laboratory and this in fact more reliable in that respect. We see from this formula that with every sample we can meet a different number of measurements. For one sample we can make maybe two with the other one five etcetera. And this is fully okay these different standard deviations then will be taken into account by different weights. So all these n minus ones here can be regarded as different statistical weights. If however, the measurement was organized in such a way that the number of measurement with each of the samples was the same, then the formula becomes simpler and the numbers of measurements can be omitted all together. I mentioned that both repeatability and within lab long-term reproducibility can be evaluated with pooled standard deviation. And let us see now how to set such experiments up in practice. | Comme nous l'avons déjà vu, chaque fois que nous voulons déterminer la fidélité, c’est-à-dire la répétabilité ou la reproductibilité intra-laboratoire, il est toujours important de faire plusieurs mesures en parallèle. Et en fait, plus on fait de mesures et plus on aura une bonne estimation de la fidélité. Souvent, les analyses peuvent être assez denses. Ce qui veut dire qu'il n'est pas facile de faire beaucoup de mesures en parallèle, notament dans un court laps de temps. Dans cette situation, un outil statistique appelé écart-type groupé devient très utile pour nous. En effet, l'écart type groupé peut-être utilisé à la fois pour le calcul de la répétabilité et de la reproductibilité intra-laboratoire. La principale formule de l'écart-type groupé est ici. Dans le cas de l'écart type groupé, on réalise toujours les mesures avec plusieurs échantillons. Et dans cette formule, ces nombres ici représentent le numéro d'échantillon. Donc nous avons jusqu’à k échantillons. Et chaque s est l'écart-type de chaque échantillon. S1 correspond à l'écart type de l'échantillon 1, s2 l'écart-type de l'échantillon n°2, etc… Donc n désigne le nombre de mesures parallèles faites avec chaque échantillon. Donc, tous ces écarts-types que l'on obtient pour chaque échantillon, on les élève au carré, on les multiplie par le nombre de mesures faites en parallèle moins 1. Ces quantités sont ensuite sommées entre elles et on les divise par ce nombre, qui est en fait la somme de TOUTES les mesures faites en parallèle (sur tous les échantillons) moins le nombre d'échantillons. Et ce qu'on finit par obtenir, c'est l'écart-type groupé. Ce qui est intéressant à propos de l'écart-type groupé, c'est que vous pouvez avoir un grand nombre d' échantillons et pour chacun d'entre eux le nombre de mesures faite peut être très petit. En fait, c'est suffisant si avec chaque échantillon, au moins 2 mesures sont réalisées tant qu'il y a beaucoup d'échantillons (sur lesquels faire les mesures). Le nombre de degrés de liberté pour l'écart type groupé est en fait ce nombre ici. Donc, si nous avons disons 10 échantillons mais qu'on ne fait avec chacun d'entre eux que 2 mesures, alors ce nombre de degrés de libertés vaudra 10*2 - 10, ce qui est égal à 10. Ce qui veut dire que c'est, statistiquement parlant, environ aussi bien que 11 mesures parallèles faites sur le même échantillon. Donc, bien que chacun de ses écarts-types, qui sont obtenus à partir de 2 mesures parallèles, soient très, très, très, très peu fiables, mais si nous en avons beaucoup (d'écarts-types) et que nous les moyennons selon cette formule, alors l'écart-type groupé résultant sera assez fiable en fait. C'est très important que tous ces échantillons sur lesquels nous avons réalisé les mesures, doivent être similaires. C'est à dire que leurs écarts-types doivent être semblables et similaires. Si les échantillons ont des niveaux de concentrations différents, des matrices très différentes, ou des niveaux de difficultés de préparation d'échantillon très différents, alors l'écart-type groupé résultant ne signifiera pas grand-chose. Maintenant, si nous comparons l'écart-type qui est en fait l'écart-type groupé obtenu à partir d'un nombre différent d'échantillons dans le cas ou peu de mesures en parallèle ont été effectuées sur chaque échantillons avec le deuxième cas où on ne posséderait qu'un seul échantillon et où on ferait un grand nombre de mesure, alors, en fait, l'écart-type groupé (obtenu dans le 1er cas) peut nous donner une estimation plus fiable de la fidélité car les propriétés un peu différente de chaque échantillons seront toutes prises en compte. Ce qui fera que, l'écart-type groupé sera, effectivement, un écart-type moyen de tous les échantillons. Et de plus, souvent, ces écarts-types sont estimés sur plusieurs jours. En effet, pour un jour, peut-être que notre appareil de mesure peut marcher un peu mieux mais un autre jour, un peu moins bien. À nouveau, l'écart-type groupé résultant nous donne une moyenne des conditions expérimentales dans notre laboratoire. Si nous déterminons un écart-type sur un seul jour avec un seul échantillon, alors il peut arriver que cet échantillon en particulier soit un bon échantillon et l'appareil, sur ce jour, peut également être dans de très bonnes conditions de fonctionnement, ce qui peut alors donner une très faible incertitude type qui est également l'incertitude type de répétabilité dans ce cas. Cela donne 2 "bonnes" incertitudes pour de futures applications car lorsque nous déterminons ces écarts-types, nous le faisons afin de pouvoir les réutiliser plus tard, dans le temps, pour des calculs d'incertitudes de mesurages. Alors que l'écart-type groupé, nous donne une situation moyenne de notre laboratoire ce qui est en fait plus fiable. Nous voyons, sur cette formule qu'avec chaque échantillon, nous pouvons avoir un nombre différent de mesures. Pour un échantillon, on peut faire peut-être 2 mesures, mais avec un autre plutôt 5 mesures, etc… Et cela est totalement valide car les différents écarts-types obtenus seront ensuite pondérés par différents poids. Donc tous ces n moins 1 (n-1) ici peuvent être vu comme différents poids statistiques. Si, cependant, les mesures étaient organisées de telle manière que le nombre de mesures de chaque échantillons soit le même, alors la formule devient plus simple et le nombre de mesures de chaque échantillon peut se factoriser. J'ai évoqué plus tôt que la répétabilité et la reproductibilité intra-laboratoire peuvent être évaluées à l'aide de l'écart-type groupé. Regardons maintenant comment mettre en pratique cet objectif. |
Selon la façon dont les expériences sont planifiées, l'écart type groupé peut être utilisé pour calculer la répétabilité sr ou la reproductibilité intra-laboratoire sRW.
Le plan expérimental et les calculs lors de la recherche de répétabilité sr sont expliqués dans la vidéo suivante:
Ecart type groupé en pratique : estimation de la répatabilité
http://www.uttv.ee/naita?id=18232
| I mentioned that both repeatability and within lab long-term reproducibility can be evaluated with pooled standard deviation, and let us see now how to set such experiments up in practice. Let us look now how the experimental logic will look like if we want to estimate repeatability from pooled standard deviation. I will explain this with a graph. On one axis of the graph, we will have samples, and on the other axis we have days. And suppose we have four different samples. One, two, three, four. And now since with each of the sample, for correct repeatability estimation we have to carry out the full analytical procedure including also the sample preparation, then we cannot make many many many measurements within a single day. But suppose on day one we work with sample number one and we make all together four measurements with it. One, two, three, four. On day two, we work with sample number three and we make three measurements with it. Let me also mark the days here. On day three we will work with sample number two, and we will make five mesurements. And finally on day four we will work with sample number four, and we will make two measurements. So from each of these sets of measurements, we can calculate repeatability for this particular sample on this particular day. Of course, obviously these days do not need to be consecutive days, they can be far apart from each other separated by several weeks. Now, in this particular case n1 will be 4, n2 will be 5, n3 will be 3 and n4 will be 2. And if we add all these numbers up, we will get : 4 plus 5 equal to 9, plus 3 equal to 12, plus 2 equal to 14, and if we subtract from this the number of samples, which is 4, we get 10 which will be for us the number of degrees of freedom. Meaning the pooled standard deviation calculated by this approach, corresponds to the situation as if we had made 11 measurements within a single day on one single sample. But as I explained previously, this approach, in fact, gives you a more reliable repeatability estimate. It goes different sample envolved which can be slightly different, by their matrix or by the difficulty of analysis, and also different days are involved, meaning the laboratory settings can be slightly different. Let us look now at the practical calculation of pooled standard deviation from the example of determination of an antibiotic, meropenem, in blood plasma by HPLC. And here are the data which exactly correspond to the situation which I just showed on the whiteboard. So different days, different samples and different numbers of parallel measurements. And I have also incorporated here into the slide, the formula by which the pooled standard deviation is calculated. And we will first start by calculating the standard deviations for all the samples and also by writing the numbers of n-1 for all the samples. So standard deviation expressed in µg/g. And I will mark the area, in such a way that it would be suitable for each and every of this column. Those empty cells will be omitted by the STDEV function so they will not be considered by test zeros so the calculations will be correct So these are now the individual standard deviations. Now it's useful to write here the n-1 values. In this case it's 3, in this case it's 2, in this case it's 4, in this case it's 1. And now it's useful to make a row where the n-1 is multiplied by the standard deviation squared. n-1 multiplied by s squared. So… Now we have all the data needed for calculating the pooled standard deviation, and let me just comment on the denominator of this ratio here. If we sum up n1, n2, etc... and subtract then the K, then in fact this will be exactly the same as the sum of the n-1 and since we have the n-1 values nicely available here, we will in fact instead of this use the sum of n-1 values for the calculation. So it's a repeatability pooled standard deviation and is equal to square root, and it now is a ratio of two sums : sum of these products here divided by the sum of all the n-1 values here. And of course, it is expressed in the same way in µg/g. So if we want to characterize this procedure by repeatability estimate, then this repeatability estimate would be most suitable, because it has been determined with different samples on different days. And it is important to mention that even though the days are different, since on each day we have the standard deviation which gives us repeatability because all the measurements were done on the same day, then all this are repeatability standard deviations and when we pool repeatability standard deviation we also get repeatability standard deviation. So even though the experiments were carried out on different days we do not get reproducibility but we get repeatability standard deviation. | J'ai mentionné que la répétabilité et la reproductibilité à long terme en laboratoire peuvent être évaluées avec l'écart-type global et voyons maintenant comment mettre en place de telles expériences en pratique. Regardons maintenant à quoi va ressembler la logique expérimentale si l'on veut estimer la répétabilité à partir de l'écart-type global. Je vais expliquer cela avec un graphique. Sur un axe du graphique nous aurons les échantillons, et sur l'autre axe nous avons les jours. Et supposons que nous ayons quatre échantillons différents. Un, deux, trois, quatre. Et maintenant puisque pour chaque échantillon, pour une estimation correcte de la répétabilité, nous devons effectuer la procédure d'analyse complète incluant la préparation des échantillons, alors nous ne pouvons pas faire beaucoup beaucoup beaucoup beaucoup de mesures dans une même journée. Mais supposons que le premier jour nous travaillons avec l'échantillon numéro un et nous faisons tous ensemble quatre mesures avec lui. Un, deux, trois, quatre. Le deuxième jour, nous travaillons avec l'échantillon numéro trois et nous faisons trois mesures avec lui. Permettez-moi aussi de marquer les jours ici. Le troisième jour, nous travaillerons avec l'échantillon numéro deux et nous allons faire cinq mesures. Et enfin, le quatrième jour, nous travaillerons avec l'échantillon numéro quatre et nous ferons deux mesures. Donc à partir de chacun de ces ensembles de mesures, nous pouvons calculer la répétabilité pour cet échantillon particulier en ce jour particulier. Bien sûr, il est évident que ces jours ne doivent pas nécessairement êtrs des jours consécutifs, ils peuvent être très éloignés les uns des autres, séparés par plusieurs semaines. Maintenant dans ce cas particulier n1 sera 4, n2 sera 5, n3 sera 3 et n4 sera 2. Et si nous ajoutons tous ces chiffres, nous obtenons : 4 plus 5 égal à 9, plus 3 égal à 12, plus 2 égal à 14, et si nous soustrayons à ceci le nombre d'échantillons, qui est de 4, que nous obtenons 10 qui sera pour nous le nombre de degrés de liberté. Cela signifie que l'écart type global calculé par cette approche, correspond à la situation où nous aurions fait 11 mesures en un seul jour sur un seul échantillon. Mais comme je l'ai expliqué précédemment, cette approche, en fait, nous fournie une estimation plus fiable de la répétabilité. Elle implique différents échantillons qui peuvent être légèrement différents, de part leur matrice ou de part la difficulté d'analyse, et aussi différents jours sont impliqués, ce qui signifie que les paramètres de laboratoire peuvent être légèrement différents. Examinons maintenant les aspects pratiques du calcul de l'écart type global à partir de l'exemple de la détermination d'un antibiotique, le méropénem, dans le plasma sanguin par HPLC. Et voici les données qui correspondent exactement à la situation que je viens de montrer sur le tableau blanc. Donc différents jours, différents échantillons et différents nombres de mesures parallèles. Et j'ai également intégré ici dans la feuille, la formule avec laquelle l'écart type global est calculé. Et nous allons commencer par calculer l'écart-type pour tous les échantillons et aussi en écrivant les valeurs de n-1 pour tous les échantillons. L'écart-type est exprimé en µg/g. Et je vais marquer la zone de manière appropriée pour chacune de ces colonnes. Ces cellules vides seront omises par la fonction STDEV, de sorte qu'elles ne seront pas pris en compte par le test zéro, de manière à ce que les calculs soient correctes. Ainsi, ce sont alors les écarts types individuels. Il est maintenant utile d'écrire ici les valeurs n-1. Dans ce cas c'est 3 dans ce cas c'est 2, dans celui-ci c'est 4, dans celui-là c'est 1.Et maintenant il est utile de faire une ligne où le n-1 moins 1 est multiplié par l'écart-type au carré. n-1 multiplié par s au carré. Donc… Nous avons maintenant toutes les données nécessaires pour calculer l'écart-type global et laissez-moi juste commenter le dénominateur de ce rapport ici. Si nous additionnons n1, n2, etc ... et soustrayons alors le K, alors en fait ce sera exactement la même valeur que la somme des n-1 et comme nous avons les valeurs de n-1 bien disponibles ici, nous utiliseront en fait à la place de ceci, la somme des valeurs de n-1 pour le calcul. Il s'agit donc d'un écart-type global de répétabilité et c'est égale à la racine carrée, et c'est maintenant un rapport de deux sommes : somme de ces produits ici divisés par la somme de toutes les valeurs de n-1 ici. Et bien entendu, il est exprimé de la même façon en µg/g. Donc si nous voulons caractériser cette procédure par une estimation de la répétabilité, alors cette estimation de répétabilité serait la plus approprié, parce qu'elle a été déterminée avec des échantillons différents sur des jours différents. Et il est important de mentionner que bien que les jours soient différents, puisque pour chaque jour, nous avons l'écart type ce qui nous donne une répétabilité car toutes les mesures ont été effectuées sur le même jour, alors tout cela est l'écart-type et quand nous globalisons l'écart type de répétabilité on obtient aussi l'écart type de répétabilité. Donc même si les expériences étaient effectuées à des jours différents, nous ne pourrions pas obtenir la reproductibilité, mais nous obtenons écart-type de répétabilité. |
Le plan expérimental et les calculs lors de la recherche de la reproductibilité sRW en laboratoire sont expliqués dans la vidéo suivante:
Ecart-type groupé en pratique : estimation de la reproductibilité à long terme en laboratoire
http://www.uttv.ee/naita?id=18234
| Let us look now, how to set up the experiment for determining within-lab long-term reproducibility from Pooled Standard Deviation. And again we will make a graph and again we will have samples on one axis, and suppose we can have 4 samples. And now on the other axis, we again have days but this time, we need more days and we need them certainly spread over a long time period. So let us take for example 9 different days. And again, we analyze each sample. But now, we do not analyze the sample on the same day but we will spread out the analysis across a number of days. And let's assume that sample number 1 was analyzed on day 1, one day 4 and on day 8. Sample number 2 was analyzed on day 2, day 4, day 6 and day 9. Sample number 3 was analyzed on day 1, day 3, day 5 and day 7. And finally, sample number 4 was analyzed on day 3, day 6, day 8. So we see now that on each day different samples were analyzed and also on each day, only one analysis was done but each of the samples got analyzed across a long time period because the time between each consecutive pair of days can be few weeks. So that this whole time period can easily be maybe a half of year or even a year. So it will really be a long-term reproducibility standard deviation. But now, with each sample again, we can make quite few measurements, no so many, but since we averaged across all these measurements, eventually our Pooled Standard Deviation statistically will be quite reliable. And again of course, the averaging, here, comes from the fact that we have somewhat different samples and obviously different days. Let us look now, how the calculation within-lab long-term reproducibility from Pooled Standard Deviation can be done practice. And we will again look at this on an example and this time it will be protein content determination by the Kjeldahl method. We see here now that we have several samples, five altogether. And each of these samples has been measured on several different days and those days are separated quite far apart from each other so that up to half a year time is involved in this determination. And here, we can see the results of those determinations that were obtained for these several sums. And again, it is useful to first of all calculate the standard deviation for each of these samples and then the "n-1" values and then the products of "n-1" to standard deviation squared. And then finally, we can calculate the Pooled Standard Deviation according to this form. So this time, we will organize these data in columns because this is more reasonable with this arrangement of data. Standard deviations in g/100g and we will also put here sample numbers. So for sample 1, it will be like this. For example 2, we'll get this one. For sample 3, like this. For sample 4, this one. And for sample 5, our standard deviation will be like this. Now "n-1", for sample 1, it's 2. Sample 2 also 2. Sample 3 also 2. Sample 4 also 2. And sample 5, it's 1. And now, these products "n-1" multiplied by standard deviation squared. And here, we can now copy and again as we saw in the case of repeatability calculation, we can now calculate the Poor Standard Deviation, a square root of the sum of all these components divided by the sum of the n minus 1 values which as I explained, will be exactly equal to this value here. So within-lab long-term reproducibility pooled standard deviation will be here, square root sum of all these components divided by the sum of all "n-1 values and the unit of course is g/100g. And this can now be used conveniently for characterizing the procedure for uncertainty estimation using the nordtest approach!. And it's a reliable standard deviation because it has been obtained over a long time period and also with different samples which means averaging. And the number of degrees of freedom in this particular case is altogether 9 which is the sum of these values. And of course, if the laboratory continues working in this field then it's very useful to collect more and more data, either to reanalyze these samples if it is still possible or to collect more data so that to make this standard deviation estimate still more reliable. | Voyons maintenant, comment mettre en place l'expérience pour déterminer la reproductibilité intra-laboratoire à long terme par l'écart-type groupé. Et encore une fois nous allons faire un graphique et nous aurons de nouveau des échantillons sur un axe, et supposons que nous puissions avoir 4 échantillons. Et maintenant sur l'autre axe, nous avons encore des jours mais cette fois, nous avons besoin de plus de jours et nous en avons certainement besoin sur une longue période. Prenons donc comme exemple 9 jours différents. Et à nouveau, nous analysons chaque échantillon. Mais désormais, nous n'analysons pas l'échantillon le même jour mais nous allons étaler l'analyse sur plusieurs jours. Et supposons que l'échantillon 1 a été analysé le jour 1, le jour 4 et le jour 8. L'échantillon numéro 2 a été analysé le jour 2, le jour 4, le jour 6 et le jour 9. L'échantillon numéro 3 a été analysé le jour 1, le jour 3, le jour 5 et le jour 7. Et finalement, l'échantillon numéro 4 a été analysé le jour 3, le jour 6, le jour 8. Nous voyons donc maintenant que chaque jour différents échantillons ont été analysés et aussi que chaque jour, seulement une analyse a été réalisée. mais chacun des échantillons ont été analysé sur une longue période car le temps entre chaque pair de jours consécutifs peut être quelques semaines. De telle sorte que cette période puisse être facilement un semestre ou même une année. Cela serait vraiment un écart-type de reproductibilité à long terme. Mais maintenant, avec chaque échantillon, de nouveau, nous pouvons faire assez peu de mesures, pas tant que cela, mais étant donné que nous faisons la moyenne de toutes ces mesures, notre écart-type groupé sera finalement assez fiable statistiquement. Et encore une fois bien sûr, la moyenne, ici, vient du fait que nous avons des échantillons quelque peu différents et évidemment de jours différents. Voyons maintenant, comment le calcul de la reproductibilité intra-laboratoire à long terme à partir de l'écart-type groupé peut être réalisé en pratique ? Et nous allons le voir dans un exemple et cette fois ce sera la détermination de la teneur en protéines par la méthode de Kjeldahl. Nous voyons ici que maintenant nous avons plusieurs échantillons, 5 au total. Et chacun de ces échantillons a été mesuré sur plusieurs jours et ces jours sont séparés assez loin les uns des autres de sorte qu'un temps d'une demie-année soit pris en compte dans cette détermination. Et ici, nous pouvons voir les résultats de ces déterminations qui ont été obtenus pour ces plusieurs sommes. Une fois encore, il est utile de d'abord calculer l'écart-type pour chacun de ces échantillons et ensuite les valeurs de "n-1" et puis le produit de "n-1" par l'écart-type au carré. Et finalement, nous pouvons calculer l'écart-type groupé en utilisant cette formule. Donc cette fois, nous allons organiser ces données dans des colonnes parce que cela est plus judicieux avec cette dispositon de données. L'écart-type en g/100g et nous pourrons mettre ici de simples nombres. Donc pour l'échantillon 1, ce sera comme cela. Pour l'échantillon 2, nous allons mettre cela. Pour l'échantillon 3, comme ceci. Pour l'échantillon 4, ceci. Et pour l'échantillon 5, notre écart-type sera comme cela. Maintenant "n-1", pour l'échantillon 1, c'est 2. L'échantillon 2 aussi 2. L'échantillon 3 aussi 2. L'échantillon 4 aussi 2. Et l'échantillon 5, c'est 1. Et maintenant, ces produits : "n-1" multipliés par l'écart-type au carré. Et ici, nous pouvons maintenant copier et de nouveau comme nous avons vu dans la case de calcul de répétabilité, nous pouvons désormais calculer l'écart-type groupé, une racine carrée de la somme de tout ces composants divisée par la somme des valeurs "n-1" qui comme je l'ai expliqué, sera exactement égal à cette valeur là. Donc l'écart-type groupé de la reproductibilité intra-laboratoire à long terme sera ici, la racine carrée de la somme de tous ces composants divisée par la somme de toutes les valeurs "n-1" et l'unité est bien sûr le g/100g. Et cela peut être utilisé convenablement pour caractériser la procédure de l'estimation des incertitudes utilisant l'approche Nordtest. Et c'est un écart-type fiable car il a été obtenu sur une longue période et aussi avec différents échantillons qui signifie une moyenne. Et le nombre de degrés de liberté dans ce cas particulier est au total 9 qui est la somme de ces valeurs. Et bien sûr, si le laboratoire continue de travailler dans ce domaine alors c'est très utile de collecter de plus en plus de données, soit pour analyser de nouveau ces échantillons si cela reste possible ou pour collecter plus de données pour faire que cet écart-type estimé reste plus fiable. |
Autoévaluation sur cette partie du cours : Test 6
https://sisu.ut.ee/measurement/measurement-1-3
***
[1] Il ne peut pas être strictement défini, combien de temps est «à long terme». Une définition approximative pourrait être: un an, c'est bien, «plusieurs mois» (au moins 4-5) est le minimum. Bien sûr, cela dépend aussi de la procédure.
[2] Les termes «reproductibilité intra-laboratoire» et «fidélité intermédiaire» sont synonymes. Le VIM (1) préfère la fidélité intermédiaire. Le manuel Nordtest (5) utilise la reproductibilité intra-laboratoire. Afin de souligner l'importance du «long terme», dans ce cours, nous appelons souvent sRW la reproductibilité à long terme en laboratoire.
***
Les diapositives de la présentation et des fichiers de calcul - avec les données initiales uniquement, ainsi qu'avec les calculs effectués - sont disponibles ici:
Ecart-type groupé - répétabilité - données initiales.xls
Ecart-type groupé - répétabilité - résolu.xls
Ecart-type groupé - reproductibilité - données initiales.xls
Ecart-type groupé - reproductibilité - résolu.xls
8. Fidélité, justesse, exactitude
https://sisu.ut.ee/measurement/7-precision-trueness-accuracy
Résumé : Les interactions entre les différents types d'erreur (aléatoires, systématiques, totaux), leurs caractéristiques de performance correspondantes (fidélité, justesse, exactitude) et les paramètres pour exprimer quantitativement ces paramètres de performance (écart type, biais, incertitude de mesure) sont expliqués dans ce chapitre.
Interrelation entre les concepts de fidélité, justesse, exactitude et incertitude de mesure
http://www.uttv.ee/naita?id=17824
| Let us look now at the Inter-relations between some of the important concepts and parameters related to accuracy, precision, throughness and measurement uncertainty. And let us look at them with the aid of this interesting scheme. It has been published by a group of people in accreditation and quality assurance in 2006. And, it is very good aid in linking these concepts together. So, let us start from this first column, the error column. As we've seen already in one of the introductory lectures : the total error or simply the error of the analysis or measurement can be viewed as composed of random and systematic error whereby random error accounts for the random effects and systematic error for the systematic effects. Now, as we saw already in the very very beginning of this course, the concept of error is an abstract concept because, for knowing the error, we should know the true value of the quantity which almost never we can know in reality. Therefore, instead of errors we usually operate with some estimates, with some parameters which can be regarded as estimates of error. And, so we move to the second column. So, the systematic error can be estimated by trueness. So, trueness is an estimate of the systematic error. For knowing trueness, we do not need to know the true value, but we need to have a reference value, whereby reference value is a value which does have uncertainty, but not very high uncertainty. We will see later on how useful this concept actually is. Secondly, the random error can be approximated or can be estimated by precision. And, there are different precision characteristics : repeatability, reproducibility. And, there are different kinds of reproducibilities, and, all of them are estimates of random error. In the sense, that in order to arrive at truly random error, we would need to make an infinite number of measurements which we never can make. So, from a finished number of measurements, we can estimate the error. And, these two rules and precision can be combined into the concept of accuracy. So, if the measurement result had good trueness and has good precision, we can say that this measurement result is accurate. Now, how do we numerically express these ? Let us now look at the last column, trueness is numerically expressed by bias, whereby bias is the difference between the reference value and the value that we obtain from the measurement. And precision, numerically is expressed by standard deviation of the repeated measurement results. And, now, it is possible to combine the estimate of bias and the precision standard deviation if they have been found in a correct way into measurement uncertainty. And, this is something that we will also be looking at if we go to the approaches for measurement uncertainty estimation. And, there will be an approach based on within-lab validation data or the so-called "nordtest approach". And, that "nordtest approach" makes direct use of this relation as is shown here. |
Regardons maintenant les interrelations entre certains concepts et paramètres importants liés à la précision, à la rigueur de la fidélité et à l'incertitude de mesure. Et observons-les avec l'aide de ce schéma intéressant. Il a été publié par un groupe de personnes en accréditation et assurance qualité en 2006. Et, cela est une très bonne piste pour lier ces concepts ensemble. Commençons donc à partir de cette première colonne, la colonne d'erreur. Comme nous l'avons déjà vu dans l'une des conférences d'introduction, l'erreur totale ou simplement l'erreur de l'analyse ou de la mesure peut être considérée comme composé d'une erreur aléatoire et systématique par laquelle l'erreur aléatoire prend en compte les effets aléatoires et l'erreur systématique. Maintenant, pour les erreurs systématiques, comme nous l'avons déjà vu au tout début de ce cours, le concept de l'erreur est un concept abstrait parce que, pour connaître l'erreur, nous devrions savoir la vraie valeur de la quantité que nous ne pouvons presque jamais connaître en réalité. Par conséquent, au lieu d'erreurs, nous opérons habituellement avec certaines des estimations avec certains des paramètres qui peuvent être considérés comme des estimations d'erreur. Et, donc nous passons à la seconde colonne. Ainsi, l'erreur systématique peut être estimée par la justesse. La justesse est une estimation de l'erreur systématique. Pour connaître la justesse, nous n'avons pas besoin de connaître la vraie valeur, mais nous devons avoir une valeur de référence, la valeur de référence étant une valeur qui présente une incertitude, mais pas très élevée. Nous verrons plus tard à quel point ce concept est réellement utile. D'autre part, l'erreur aléatoire peut être approximée ou peut être estimée par la fidélité. Et il existe différentes caractéristiques de fidélité : répétabilité, reproductibilité. Et, il existe différents types de reproductibilités, et, tous sont des estimations de l'erreur aléatoire. Dans ce sens, pour vraiment arriver à une erreur aléatoire, nous aurions besoin de faire un nombre infini de mesures que nous pouvons jamais faire. Ainsi, à partir d'un nombre fini de mesures, nous pouvons estimer l'erreur. Et, ces deux règles et la fidélité peuvent être combinées dans le concept de précision. Ainsi, si le résultat de la mesure avait une bonne justesse et a une bonne fidélité, nous pouvons dire que ce résultat de la mesure est précis. Maintenant, comment exprimons-nous cela numériquement ? Examinons maintenant la dernière colonne, la justesse est numériquement exprimée par le biais, le biais étant la différence entre la valeur de référence et la valeur que nous obtenons à partir de la mesure. Et précisément, numériquement est exprimée par l'écart-type des résultats de mesure répétés. Et, maintenant, il est possible de combiner l'estimation du biais et l'écart-type de la fidélité s'ils ont été trouvés correctement dans l'incertitude de mesure. Et, c'est quelque chose que nous regarderons également si nous passons aux approches pour l'estimation de l'incertitude de mesure. Et, il y aura une approche basée sur les données de validation en laboratoire ou la soi-disant "approche nordtest". Et, cette "approche nordtest" fait directement l'usage de cette relation comme montré ici. |
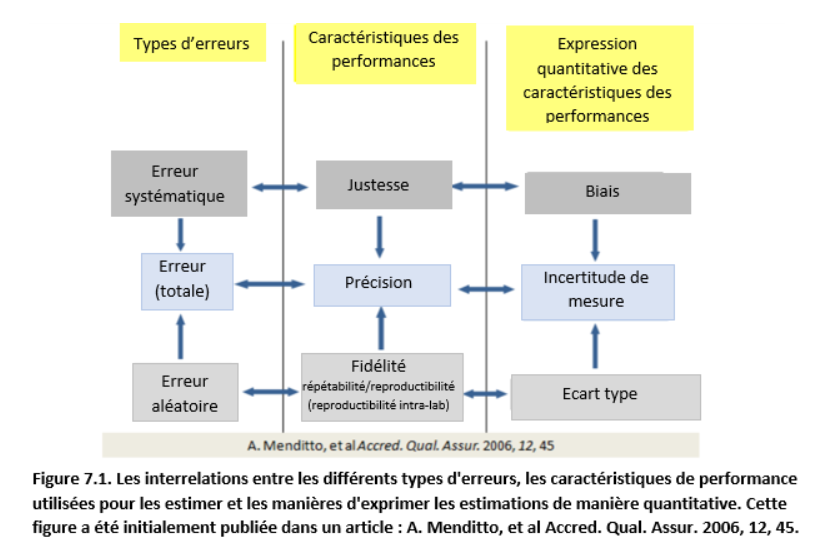
La différence entre la valeur mesurée et la valeur réelle est appelée erreur ou erreur totale (voir chapitre 1). Cette erreur peut être divisée en deux parties : erreur aléatoire (ayant une amplitude et un signe différents dans le cas de mesures répétées) et erreur systématique (ayant une amplitude et un signe identiques ou changeant systématiquement dans le cas de mesures répétées). Comme vu dans le chapitre 1, les erreurs ne peuvent pas être connues exactement. Par conséquent, au lieu des erreurs elles-mêmes, nous utilisons des estimations des erreurs - les caractéristiques de performance.
Ainsi, la justesse est l'estimation de l'erreur systématique. Pour déterminer la justesse, nous n'avons pas besoin de connaître la vraie valeur, mais nous devons connaître une valeur de référence. La valeur de référence (différente de la valeur réelle) présente une incertitude, mais généralement faible. Différents types de fidélité sont des estimations de l'erreur aléatoire. Pour obtenir la «vraie» fidélité, il faudrait effectuer un nombre infini de mesures répétées. Il existe différents types de fidélité, selon les conditions dans lesquelles la fidélité est déterminée, par ex. répétabilité (section 1) et fidélité intermédiaire (section 6). L'exactitude englobe à la fois la justesse et la fidélité et doit être considérée comme décrivant l'erreur totale.
Ces caractéristiques de performance peuvent être exprimées quantitativement.
Le biais - différence entre la valeur mesurée obtenue à partir de plusieurs mesures répétées avec le même échantillon et la valeur de référence - est l'expression quantitative de la justesse.
L'écart type - à nouveau obtenu à partir de plusieurs mesures avec le même échantillon - est l'expression quantitative de la fidélité.
Ces deux éléments peuvent être combinés en une estimation de l'incertitude de mesure, qui peut être considérée comme l'expression quantitative de l'exactitude.
Autoévaluation sur cette partie du cours : Test 7
9. L'approche par modélisation ISO GUM
https://sisu.ut.ee/measurement/9-iso-gum-modeling-approach-bottom-approach
Cette partie présente pas à pas l'approche de modélisation pour l'estimation d'incertitude de mesure. Cette approche est décrite en détail dans l'ISO GUM² et a été interprété pour la chimie dans le guide d'incertitude de mesure Eurachem³. Cette approche est souvent appelée l'approche de "bottom-up". Cela veut dire que les incertitudes des quantités d'entrée sont trouvées et ensuite combinées dans l'incertitude-type composée. L'estimation d'incertitude réalisée dans la partie 4 utilise aussi cette approche en principe. La présentation dans cette partie est basée sur un exemple particulier - la détermination d'azote d'ammonium dans l'eau.
9.1. Etape 1 - Définition du mesurande
9.2. Etape 2 - Equation du modèle
9.3. Etape 3 - Sources d'incertitudes
9.4. Etape 4 - Valeurs des données d'entrée
9.5. Etape 5 - Incertitude des données d'entrée
9.6. Etape 6 - Valeur de la grandeur de sortie
9.7. Etape 7 - Incertitude-type composée
9.8. Etape 8 - Incertitude élargie
9.9. Etape 9 - Analyse de l'incertitude obtenue
Le support utilisé dans cette partie peut être téléchargé à partir d'ici :
uncertainty_of_photometric_nh4_determination_kragten_solved~1.xls
10. Aperçu des approches d'estimation de l'incertitude de mesure
verview-approaches-estimating-measurement-uncertainty" target="_blank">https://sisu.ut.ee/measurement/verview-approaches-estimating-measurement-uncertainty
Résumé : Dans cette partie, les principales approches qui peuvent être utilisé pour l’estimation des incertitude de mesure sont présentées.
Aperçu des approches d'estimation de l'incertitude de mesure
http://www.uttv.ee/naita?id=17704
| We have now covered most of the basic concepts and basic knowledge on measurement uncertainty estimation and we can now make a small overview of the approaches that exist for measurement uncertainty estimation And let us start from the basic question that analytical laboratory usually has when uncertainty estimation is considered. So usually lab people more or less know to uncertainty sources of the procedures because they are professionals and they use those procedures in their everyday work. Secondly they usually had lots of data of different kind they have controlled charts and proficiency testing results, parallel measurement data etc. They also have calibration data instruments, they have data of the volumetric equipment And the main question is : how to use these data to take these uncertainty sources under count. So how could a person in a laboratory make maximum use of the data that already exists to take all necessary sources of uncertainty to account and carry out the uncertainty evaluation. And there are different approaches that offer different solutions to this problem. Let us look here scheme which summarizes in very general way the main approaches that exist. So we can see that all starts from definition of the measurement. So the measurement always needs to be defined clearly whichever the approaches that we use. And then the approaches are split into single lab and inter laboratory approaches. And let us first have a look at the single lab approaches. They are in turn separated into model based and not model based approaches. And now, model-based approach is the so called modeling or sometimes also called the iso gum modeling approach which is pretty much the standard approach for measurement uncertainty evaluation especially physical science. And this approach involves component by component evaluation of uncertainties meaning the measurement procedure is looked at very carefully, different uncertainty sources and uncertainty components are found and separately quantified. There's an alternative approach which is the so-called single and validation approach. This is not the model based in the sense that although you need the model for calculating your analysis result but you don't really need the model for calculating uncertainty. This uncertainty evaluation approach takes into account more general uncertainty contributions It is not as detailed it works with general uncertainty contributions such as within lab reproducibility, laboratory bias etc. This approach has been formalized in several guides but perhaps the best known is the so-called "nord test" uncertainty guide which also we will examine more closely in this course. We could say that these two are the main approaches and, of course, whenever possible, one would perhaps consider this approach because it gives more information but it also needs much more competence and it unfortunately does have the roll back that sometimes underestimated uncertainty. This one, on the other hand, is very suitable for routine laboratories it does not need so much extra work and makes very good use of the already existing data and usually does not lead to underestimated uncertainties in fact it sometimes leads to overestimated uncertainties and it does not need as high competence level as this uncertainty estimation approach, but of course, from this approach you will not learn nearly as much about your procedures as from this approach. these are the two ones that would be recommended for using in your everyday worlk. Now, let us turn to this The inter laboratory approach. interlaboratory approaches differ from these two in the sense that actually data from your lab, or situation from your lab is not taken into account at all in any way. And this is why these approaches are not recommended. In fact the main use of these approaches would be to find out more or less what the uncertainty could be in the situation that you even don't yet have the procedure and you want to know preliminarily what you can expect from this procedure. So in routine everyday work, these approaches are not recommended and we will not touch deeply these approaches also in this course. The approaches that we looked at they give uncertainties that refer to different situation. So this modeling approach in fact calculates the uncertainty for the individual actual result that you had on one concrete individual day with one concrete sample. The single lab validation approach calculates a typical uncertainty of the results which you can obtain with the particular procedure so that this uncertainty estimate is not directly linked to any particular sample. It's rather linked to a procedure while this one can be linked to a real particular sample or real individual sample. And now, this interlaboratory approaches in fact gives some typical general uncertainty which can be expected from this procedure when used in different level. | Nous avons maintenant couvert la plupart des concepts de base et des connaissances de base sur l'estimation de l'incertitude de mesure et nous pouvons maintenant faire un petit aperçu des approches qui existent pour l'estimation de l'incertitude de mesure. Et commençons par la question de base que le laboratoire d'analyse se pose généralement quand l'estimation d'incertitude est considérée. Donc, généralement, les personnels de laboratoire connaissent plus ou moins les sources d'incertitude des procédures car ce sont des professionnels et qu'ils utilisent ces procédures dans leur travail quotidien. Deuxièmement, ils ont généralement beaucoup de données de différents types, ils ont des diagrammes contrôlés, et des résultats de tests de compétence, des données de mesures parallèles etc. Ils ont aussi des données des instruments d'étalonnage, ils ont des données de l'équipement volumétrique, et la principale question est : comment utiliser ces données pour prendre en compte ces sources d'incertitude. Alors, comment une personne dans un laboratoire pourrait-elle utiliser au maximum les données qui existent déjà pour prendre en compte toutes les sources d'incertitudes nécessaires et effectuer l'évaluation de l'incertitude. Et il y existe différentes approches qui offrent différentes solutions à ce problème. Regardons ici un schéma qui résume d'une façon très générale les principales approches qui existent. Nous pouvons donc voir que tout commence par la définition d'une mesure. La mesure doit donc toujours être clairement définit quelles que soient les approches que nous utilisons. Et ensuite les approches sont divisées en approches de laboratoire unique et approche inter laboratoire. Et voyons d'abord les approches de laboratoire unique. Elles sont à leur tour divisées en approches basées sur un modèle et non basée sur un modèle. Et maintenant, l'approche basée sur un modèle est aussi appelée modélisation ou parfois également appelées approche de modélisation ISO GUM ce qui est à peu près l'approche standard pour l'évaluation de l'incertitude de mesure en particulier pour les sciences physiques. Et cette approche implique une évaluation composante par composante de l'incertitude, ce qui signifie que la procédure de mesure est examinée très attentivement, différentes sources d'incertitude et composantes d'incertitude sont trouvées et quantifiées séparément. Il y a une approche alternative qui est appelée approche de validation en laboratoire unique. Ce n'est pas basée sur un modèle, dans le sens où, bien que vous ayez besoin d'un modèle pour calculer vos résultats d'analyse, vous n'avez pas vraiment besoin d'un modèle pour calculer l'incertitude. Cette approche d'évaluation de l'incertitude prend en compte des contributions à l'incertitude plus générales. Elle n'est pas aussi détaillée, elle fonctionne avec des contributions d'incertitude plus générales telle que la reproductibilité en laboratoire, le biais de laboratoire etc. Cette approche a été formalisée dans plusieurs guides mais peut être le plus connu est le guide du test d'incertitude "Nord" que nous examinerons également de plus près dans ce cours. Nous pourrions dire que ces deux approches sont les principales et, bien sûr, dans la mesure du possible, on pourrait peut-être considérer cette approche car elle donne plus d'informations mais elle nécessite beaucoup plus de compétences et elle possède un recul qui parfois sous-estime l'incertitude. Celle-là, d'autre part, est très appropriée pour les laboratoires de routine, elle ne nécessite pas trop de travail supplémentaire et permet un très bon usage des données déjà existantes et ne conduit généralement pas à une sous-estimation des incertitudes, en fait elle peut parfois conduire à une surestimation des incertitudes. Et elle ne nécessite pas un si haut niveau de compétence que cette approche d'estimation d'incertitude, mais bien sûr, à partir de cette approche, vous n'en apprendrez pas autant sur vos procédures qu'avec cette approche. Ce sont les deux approches qui seraient recommandées pour une utilisation dans votre travail quotidien. Passons maintenant à cette approche inter laboratoire. Les approches inter laboratoire diffèrent des deux autres dans le sens où les données de votre laboratoire, ou la situation de votre laboratoire ne sont en aucun cas prises en compte. Et c'est pourquoi ces approches ne sont pas recommandées. En fait, l'utilisation principale de ces approches serait de découvrir plus ou moins quelle pourrait être l'incertitude dans la situation où vous n'avez même pas encore la procédure et que vous voulez savoir au préalable ce que vous pouvez attendre de cette procédure. Donc dans le travail quotidien de routine, ces approches ne sont pas recommandées et nous n'aborderons pas non plus profondément ces approches dans ce cours. Les approches que nous avons regardées donnent des incertitudes qui se réfèrent à différentes situations. Donc cette approche de modélisation calcule en fait l'incertitude pour le résultat réel individuel que vous avez eu sur un jour individuel concret avec un échantillon concret. L'approche de validation en laboratoire unique calcule une incertitude typique des résultats que vous pouvez obtenir avec une procédure particulière afin que cette estimation d'incertitude ne soit pas directement liée à un échantillon particulier. Elle est plutôt liée à une procédure alors que celle-ci peut être liée à un véritable échantillon particulier ou un véritable échantillon individuel. Et maintenant, cette approche inter laboratoire donne en fait une certaine incertitude générale typique qui peut être attendue de cette procédure quand elle est utilisée à différents niveaux. |
Les deux approches principales qui sont abordées dans ce cours – la modélisation (ISO GUM) et l’approche de validation intra-laboratoire (Nordtest) diffèrent par leur quantité de détails, par la façon dont les effets aléatoires sont pris en considération et par le statut de l’incertitude obtenue. La table 8.1 nous résume ces différences. A voir également, Table 11.1 dans la partie 11.
Table 8.1 Quelques différences clés entre les approches de modélisation et validation intra-laboratoire pour la mesure d’estimation d’incertitude.
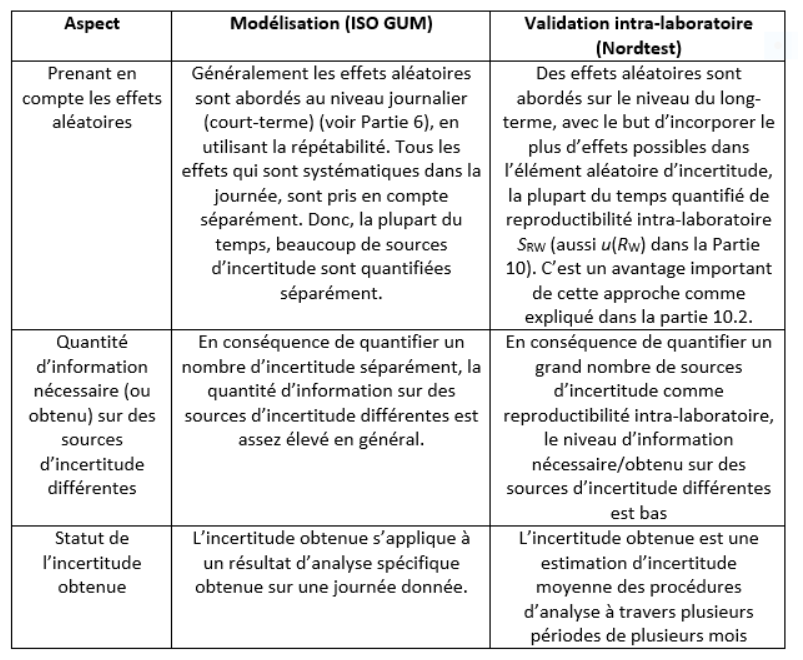
Bien que ISO GUM(2) soit le document de facto standard de l’estimation d’incertitude de mesure, particulièrement en physique, l’approche de validation intra-laboratoire (Nordtest) gagne aussi en popularité. Par exemple, l’approche Nordtest est utilisée comme référence pour la fondation de l’ISO 11352 concernant les normes applicables à la qualité de l'eau [1].
Autotest portant sur cette partie du cours : Test 8
https://sisu.ut.ee/measurement/node/1261
***
10.1. Définition du mesurande
https://sisu.ut.ee/measurement/91-measurand-model-sources-uncertainty
Définition du mesurande
http://www.uttv.ee/naita?id=17636
| We now have most of the knowledge that we need for measurement uncertainty estimation. And let us now try to apply this to a more serious, a more complex example, an example that would be quite like a real life chemical analysis. And, as this example, I have chosen determination of ammonium ion in water. Ammonium ion is an analyte which is very often determined in water, also in drinking water, in swimming pool water, etc … And it is one of important characteristics of water quality. And, very often, ammonium ion determination is carried out in spectrophotometric method, and this is exactly what we will be looking at also. In the essence of the method is the following ammonium ion itself does not have any absorbance in spectrophotometry, and this is quite common with chemical analysis that the analyte itself does not absorb, but it is converted to a dye, or to a photometric complex. And this photometric complex then has intensive absorbance in the visible region and this intensity of this absorbance is measured. The higher the intensity, the higher the concentration of ammonium ion. And since spectrophotometer by itself does not know which absorbance corresponds to which concentration of ammonium ion, then a calibration graph is constructed, and then the concentration of ammonium ion, expressed as ammonium nitrogen, is found from the calibration graph. And we will now look at this determination and the uncertainty estimation in a step-by-step manner. And we will first start with the measurement procedure. So there are two part In it first of all, we have a sample. The sample is diluted to achieve a suitable concentration, and then a photometric reaction is carried out with the photometric reagent, so that the dye is formed. And then the absorbance, the intensity of absorbance of the dye is measured with the spectrophotometer. And secondly, a calibration graph is composed using several solutions, with known concentrations of a morning. And from that calibration graph then, the eventually the ammonium nitrogen content, in the real sample, is calculated. You can see here symbols of different quantities and, in few slides, it will become evident why they are here. And these are all the quantities which will be appearing in our measurement model. And now, this modelling approach, for measurement uncertainty estimation, has a number of steps, and the step number one is measurand definition. And measurand, as we know, is the quantity that is intended to be measured. It may seem that it is very easy to define the measurand indeed in this case and, in fact, it indeed is not really difficult. But there are a couple of things that need to be taken into account. First of all, we can express the concentration as ammonium ion concentration, or we can also express it as just ammonium nitrogen concentration. And in water, studies it is more common to express ammonium concentration, not as ammonium ion but as ammonium nitrogen concern content in the sample, expressed in mg/L. So that how many milligrams of ammonium nitrogen is there in 1 liter of the water ? And our sample is a water sample, and in this case we also measure the ammonium ion concentration in the sample, not in the whole overall object. Meaning the sampling uncertainty, for us, will not be affected. | Nous avons maintenant la plupart des connaissances dont nous avons besoin pour l'estimation de l'incertitude de mesure. Et maintenant nous allons essayer d'appliquer cette estimation à un exemple plus sérieux, plus complexe, un exemple qui pourrait être assez semblable à une réelle analyse chimique. Et, pour cet exemple, j'ai choisi la détermination des ions ammoniums dans l'eau. L'ion ammonium est un analyte qui est très souvent déterminé dans l'eau, également dans l'eau potable, dans l'eau de piscine, etc ... Et c'est l'une des caractéristiques importantes de la qualité de l'eau. Et, très souvent, la détermination de l'ion ammonium est mise en œuvre par méthode spectrophotométrique, et c'est exactement ce que nous allons voir aussi. L'essence de la méthode est que l'ion ammonium suivant n'a aucune absorbance en spectrophotométrie, et c'est assez commun avec des analyses chimiques que l'analyte n'absorbe pas, mais il est converti en un colorant, ou en un complexe photométrique. Ce complexe photométrique a alors une absorbance intense dans la région du visible et l'intensité de cette absorbance est mesurée. Plus l'intensité est élevée, plus la concentration en ion ammonium est élevée. Et puisque le spectrophotomètre ne sait pas quelle absorbance correspond à telle concentration en ion ammonium, alors une droite d'étalonnage est tracée, puis la concentration en ion ammonium, exprimée en azote sous forme d'ammonium, est trouvée en utilisant la droite d'étalonnage. Nous allons maintenant regarder cette détermination et l'estimation de l'incertitude étape par étape. Nous commencerons avec la procédure de mesure. Il y a donc deux parties. En premier lieu, nous avons l'échantillon. L'échantillon est dilué pour obtenir une concentration acceptable, puis une réaction photométrique est mise en œuvre avec un réactif photométrique, pour que le colorant soit formé. Puis l'absorbance, l'intensité d'absorbance du colorant est mesurée avec la spectrophotomètre. Dans un second temps, une droite d'étalonnage est tracée en utilisant plusieurs solutions, de concentrations connues. De cette droite d'étalonnage, la teneur éventuelle en azote ammoniacal, dans l'échantillon, est calculée. Vous pouvez voir ici les symboles des différentes quantités et, en quelques diapositives, il deviendra évident de la raison pour laquelle ils sont ici. Ce sont toutes les quantités qui apparaîtront dans notre modèle de mesure. Maintenant, cette approche de modélisation, pour l'estimation de l'incertitude de mesure, a un nombre d'étapes, et la première étape est la définition du mesurande. Le mesurande, comme nous le savons, est la quantité qui est destinée à être mesurée. On peut penser qu'il est très facile de définir le mesurande dans ce cas et, en fait, ce n'est pas vraiment difficile. Mais il y a quelques éléments qui doivent être pris en compte. En premier lieu, nous pouvons exprimer la concentration en concentration d'ion ammonium, ou nous pouvons également l'exprimer en concentration d'azote sous forme d'ammonium. Et dans l'eau, il est plus courant d'exprimer la concentration d'ammonium, non pas en tant qu'ion ammonium mais en tant que teneur en azote sous forme d'ammonium dans l'échantillon, exprimée en mg/L. Alors combien de milligrammes d'azote sous forme d'ammonium y a-t-il dans un litre d'eau ? Notre échantillon est un échantillon d'eau, dans ce cas nous mesurons aussi la concentration en ion ammonium dans l'échantillon, et non dans son intégralité. Ce qui signifie que l'incertitude d'échantillonnage, pour nous, ne sera pas affectée. |
La définition du mesurande est l'étape la plus fondamentale de toutes mesures. Dans cette étape, il est défini ce qui est réellement mesuré et cette définition est également la base de la procédure de mesure et de l'équation du modèle.
Dans ce cas, le mesurande est la concentration de NH4+ exprimée en concentration d'ion ammonium CN_échantillon en [mg/l] dans l'échantillon d'eau.
10.2. Equation du modèle
Équation du modèle
http://www.uttv.ee/naita?id=17637
| Let us now move on. Step number 2 is the model, for the model equation as it is often said. Model is the mathematical expression which connects the output quantity, or the measurand, or the quantity that we intend to measure with different input quantities, whereby the input quantities are the quantities that are measured directly. And, in our case, input quantities are different kinds of absorbance values, they are volumetric data, and, also the slope and intercept of the calibration graph. And, if we now compile this model into a mathematical expression and, it will be looking like this. Asample here is the absorbance of the sample solution. And, b0 and b1 are the intercept, and the slope of the calibration graph respectively. And, actually, if we take just this part of the model equation, then this part will exactly give us the content of ammonium nitrogen in the solution that was made from the sample. So, sample is diluted before measurement. And, if we now want to extend this to the whole sample, not just the diluted sample, we have to multiply this by the dilution factor. And, it is fair to say that, just for calculating the result, this part of the model is, in fact, sufficient because the value that is given by this part of the model really will be our resulting value. However, we need to include one more term into this model, and, in fact, this ΔCdc is an additional term which carries value zero as we will see afterwards, but which has uncertainty. And, via this term, we take into account some uncertainty sources, most importantly the uncertainty due to possibility composition or contamination of the sample. | Passons maintenant à l'étape suivante. L'étape numéro 2 est le modèle, pour l'équation du modèle comme on le dit souvent. Le modèle est l'expression mathématique qui relie la quantité de sortie, le mesurande, ou la quantité que nous avons l'intention de mesurer avec différentes quantités d'entrées, les quantités d'entrées étant les quantités qui sont mesurées directement. Et dans notre cas, les quantités d'entrées sont différents types de valeurs d'absorbance, ce sont des données volumétriques ainsi que la pente et l'ordonnée à l'origine du graphique d'étalonnage. Et si nous compilons maintenant ce modèle en une expression mathématique, il ressemblera à ceci. A(échantillon) est ici l'absorbance de la solution de l'échantillon. Et b0 et b1 sont l'ordonnée à l'origine et la pente du graphique d'étalonnage respectivement. Et si nous prenons seulement cette partie de l'équation du modèle, alors cette partie nous donnera exactement la teneur d'azote d'ammonium dans la solution qui a été faite à partir de l'échantillon. Ainsi, l'échantillon est dilué avant la mesure. Et, si nous voulons maintenant étendre cela à l'ensemble de l'échantillon, pas seulement à l'échantillon dilué, nous devons le multiplier par le facteur de dilution. Et, il est juste de dire que, juste pour calculer le résultat, cette partie du modèle est, en fait, suffisante parce que la valeur qui est donnée par cette partie du modèle sera vraiment notre valeur résultante. Cependant, nous devons inclure un terme supplémentaire dans ce modèle, et ce ΔCdc est un terme additionnel qui porte la valeur zéro comme nous le verrons plus tard, mais qui a une incertitude. Et, par ce terme, nous prenons en compte certaines sources d'incertitude, surtout l'incertitude due à la possibilité de composition ou de contamination de l'échantillon. |
L'équation du modèle (équation 9.1) permet de calculer la valeur de quantité de sortie (valeur de résultat) à partir des valeurs de quantité d'entrée . Les grandeurs d'entrée sont les grandeurs directement mesurées (ou sont calculées à partir des grandeurs directement mesurées). De plus, l'équation du modèle doit permettre de tenir compte de toutes les sources importantes d'incertitude de mesure. Dans le cas de cette analyse, l'équation du modèle est la suivante:
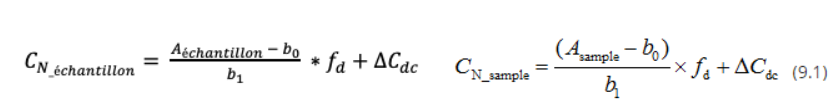

La quantité de sortie est CN_échantillon - concentration d'ions ammonium dans l'échantillon d'eau.
Les quantités entrées sont:
Aéchantillon/sample - absorbance de la solution de colorant obtenue à partir de l'échantillon;
b1 et b0 - pente et ordonnée à l'origine du graphique d'étalonnage;
fd - facteur de dilution;
ΔCdc - composant tenant compte de l'incertitude provenant d'une décomposition ou d'une contamination possible.
Les paramètres Aéchantillon , b1 , b0 et fd dans l'équation représentent les grandeurs d'entrée directement mesurées. En revanche, ΔCdc n'est introduit que pour tenir compte de l'incertitude due à une éventuelle décomposition du complexe photométrique et à une éventuelle contamination. Sa valeur est nulle comme on le verra dans la section 9.4, donc, elle ne contribue pas à la valeur de CN_échantillon . Cependant, son incertitude est différente de zéro et contribuera donc à l'incertitude de CN_échantillon.
Tests de positionnement
10.3. Sources d'incertitudes
https://sisu.ut.ee/measurement/93-practical-example
Toutes les sources d'incertitude possibles doivent être prises en compte et celles qui sont susceptibles d'avoir une influence doivent être prises en compte. Les schémas de cette section montrent les sources d'incertitude qui influencent le résultat de cette analyse. Veuillez noter, cependant, que tous ne doivent pas nécessairement être pris en compte individuellement dans l'estimation de l'incertitude. Certains peuvent être petits pour pouvoir être négligés. Certains peuvent être regroupés en une seule composante d'incertitude.
Sources d'incertitudes : en général
http://www.uttv.ee/naita?id=17638
| Let us now look further. The next step is that we should consider and identify all uncertainty sources. And those uncertainty sources that are important that have high weight in our resulting uncertainty those need to be taken into account. And this can be done either individually or by grouping them. And as we will see later on this grouping approach also is used in this example. And importantly each uncertainty source has to be linked with some quantity in the model. So that in fact this input quantity in the model is specifically brought in to link it with certain uncertainty sources which otherwise would remain unaccounted for. And if we discover an important uncertainty source that cannot be linked to any quantity in the model, then we need to modify our model. So that in fact this model that we have here can already be regarded as a modified model. The initial model would be like this and the modification that was added can be seen here. | Allons plus en détail. La prochaine étape est que nous devrions considérer et identifier toutes les sources d'incertitudes. Ces sources d'incertitudes, qui sont importantes et qui ont une grande influence sur nos incertitudes de résultats, sont à prendre en compte. Cela peut se faire individuellement ou en les groupant. Et comme nous le verrons plus tard, cette approche de regroupement est aussi utilisée dans cet exemple. Très important, chaque source d'incertitude doit être reliée à des données dans ce modèle. Donc, cette donnée d'entrée de ce modèle est spécifiquement apportée pour relier cela avec certaines sources d'incertitude qui autrement resteraient non prises en compte. Si l'on découvre une source d'incertitude importante qui ne peut être reliée à aucune donnée de notre modèle, alors nous devons modifier notre modèle. Donc, ce modèle que nous avons ici peut déjà être considéré comme modifié. Le modèle initial serait comme ceci et les modifications qui ont été ajoutées sont visibles ici. |
Sources d'incertitudes : cas par cas
http://www.uttv.ee/naita?id=17639
| Let us now look at the uncertainty sources one by one. So I have grouped them on this and the following slide according to the categories of the chemical analysis procedure. And chemical analysis starts with sampling. And since our measurement is only related to the sample not to the overall object then sampling uncertainty for us is not an uncertainty source and we do not need to take it into account. But the next step a very important one is sample preparation. And this has numerous factors that affect it. First of all the sample can be inhomogeneous the analyte separation from the sample can be incomplete and also the analyte can adsorb on the vessel walls. Luckily, in this case we have a homogeneous sample, water sample. We do not separate the analyte from the sample so there's no analyte separation effect and also, ammonium ion does not really adsorb on vessel walls. But the sample preparation has other uncertainty sources which are important. The analyte, or the photometric complex can decompose and this is certainly a factor also in ammonia determination because the photometric complex indeed loses its color with time. The analyte can in principle volatilize, with ammonium it's not very serious but still can happen a little bit. The reaction with the photometric reagent can be incomplete. And this uncertainty source actually is coupled to this one because from the point of view our result, it's not important whether the photometric complex partially decomposed or it did not even for. So these two effects actually have the same effect eventually on the result. And finally, contamination is very much possible with ammonium determination because ammonium is everywhere around us. And all these factors listed here are taken into account by this additional term ∆Cdc which covers the additional factors that can influence the analysis result. And now the next uncertainty sources are listed here. Preparation and dilution of solutions here. It is first of all kept in mind the sample solution and this is accounted for by the dilution factor fd. Then weighing comes into play with preparation of the calibration solutions because the standard substance has to be weigh. The sample we measured by volume a in the case of sample amount weighing and it's not taken into account. And the weighing in the case of calibration craf is taken into account by b1 and b0. Then calibration of the photometer. This has inturn two uncertainty sources : the purity of the standard substance and also the preparation of solutions. And both of these uncertainty sources have again effect on the calibration graph slope Finally, the measurement of the sample itself meaning the photometric measurement with the sample solution. There can be interferences from other components in the sample that also can absorb radiation and that can give us increased results. The repeatability of the reading is of course a factor meaning that if we measure the same solution absorbance several times we get slightly different results. And finally drift of the reading this means that if we measure with some time lag then we can get systematically different results because the property of the spectrophotometer slightly change over time. And all these effects are taken into account by the input quantity Asample, the sample absorbance. And then finally we have here memory effects and memory effects in the case of ammonium determination do not come into play. | Intéressons-nous maintenant aux sources d'incertitudes une à une. Je les ai regroupé ici et sur la diapositive suivante en fonction des catégories des procédures de l'analyse chimique. L'analyse chimique commence avec l'échantillonnage. Puisque la mesure est seulement liée à l'échantillon, pas à l'objet total, alors l'incertitude d'échantillonnage, pour nous, n'est pas une source d'incertitude et nous n'avons pas à la prendre en compte. La prochaine étape très importante est la préparation d'échantillon. Elle a de nombreux facteurs qui affectent l'incertitude. Premièrement, l'échantillon peut être inhomogène, la séparation de l'analyte de l'échantillon peut être incomplète et l'analyte peut s'adsorber sur les parois de la verrerie. Heureusement, dans ce cas, nous avons un échantillon homogène, de type aqueux. Nous n'avons pas à séparer l'analyte de l'échantillon donc il n'y a pas d'effet lié à la séparation de l'analyte et enfin, l'ion ammonium ne s'adsorbe pas vraiment sur les parois de la verrerie. La préparation d'échantillon a d'autres sources d'incertitudes qui sont importantes. L'analyte, ou le complexe photométrique peut se décomposer et c'est biensûr un facteur (d'incertitude) dans la détermination d'ammonium car le complexe photométrique perd de sa couleur avec le temps. L'analyte peut se volatiliser, avec l'ammonium ce n'est pas tellement le cas mais cela peut quand même arriver un peu. La réaction avec le réactif photométrique peut être incomplète. Cette source d'incertitude est enfait couplée avec celle-ci car du point de vue de nos résultats, il n'est pas important que le complexe photométrique se décompose partiellement ou qu'il n'y en ait pas de formé. Ces deux effets, ont enfait le même effet éventuellement sur le résultat. Enfin, la contamination est très plausible avec la détermination d'ammonium car l'ammonium est présent partout. Tous ces facteurs listés ici doivent être pris en compte via ce terme additionnel ∆Cdc qui couvre les facteurs additionnels qui peuvent influencer le résultat de l'analyse. Les prochaines sources d'incertitudes sont listées ici. La préparation et la dilution des solutions ici. Gardez en tête que c'est avant tout la solution échantillon et ceci est comptabilisé par le facteur de dilution fd. La pesée entre aussi en jeu avec la préparation des solutions d'étalonnage car le composé standard doit être pesé. Nous mesurons l'échantillon ici en volume et le cas de la pesée d'échantillon ici n'est pas pris en compte. La pesée, dans le cas du bon étalonnage, est pris en compte par b1 et b0. Ensuite, l'étalonnage du photomètre. Ce dernier possède deux sources internes d'incertitude : la pureté du composé de référence et la préparation des solutions. Ces deux sources d'incertitude ont aussi un effet sur la pente du graphique d'étalonnage. Enfin, la mesure de l'échantillon même, c'est-à-dire la mesure photométrique avec la solution échantillon. Il peut y avoir les interférences des autres composés dans l'échantillon qui peuvent aussi absorber la radiation et nous donner un résultat exagéré. La répétabilité de lecture est biensûr un facteur c'est-à-dire que si nous mesurons la même absorbance pour la solution plusieurs fois, nous obtenons une petite différence dans les résultats. Finalement, la dérive de lecture c'est-à-dire que si nous mesurons avec un temps de décalage alors on peut avoir systématiquement des résultats différents car les propriétés du spectrophotomètre changent un peu dans le temps. Tous ces effets sont pris en compte par la donnée d'entrée Aéchantillon, l'absorbance de l'échantillon. Pour finir, nous avons des effets de mémoire et dans le cas de l'ammonium, ils ne sont pas à prendre en compte. |
Les schémas suivants répertorient les principales sources d'incertitudes dans l'analyse chimique et commentent la présence ou l'absence des sources d'incertitudes respectives dans notre cas. [1]
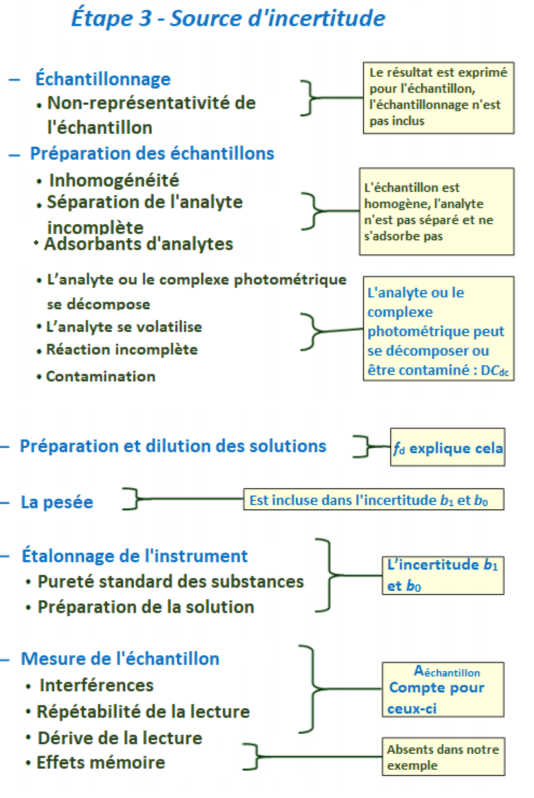
L'influence des sources d'incertitudes de mesure, regroupées en fonction des grandeurs d'entrée, sur le résultat peut être schématisée sous la forme du diagramme dit «arête de poisson» :
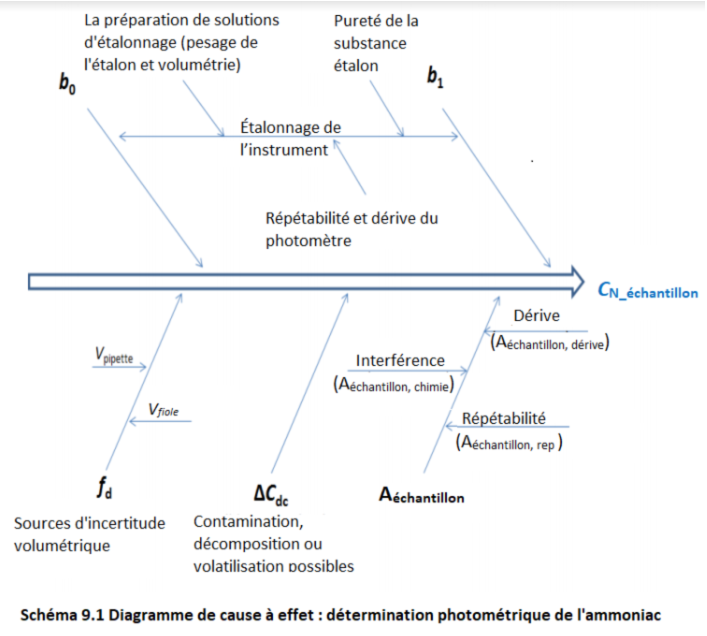
A propos de la modification du modèle
Comme expliqué dans la vidéo, cette équation de modèle,

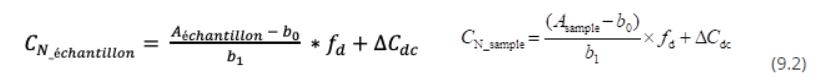
identique à l'équation 9.1 (section 9.2) peut être considérée comme un modèle modifié : le terme ∆Cdc est utilisé pour tenir compte de certaines sources d'incertitudes (décomposition possible, contamination éventuelle). Ce terme est un terme additif , c'est-à-dire que son influence sur C N_échantillon est constante, quelle que soit la valeur de C N_échantillon . La valeur de ∆Cdc est de 0 mg/L et son incertitude exprime l' absolu (c'est-à-dire exprimée en mg/L) incertitude correspondant aux effets possibles de la décomposition et de la contamination. L'intégration de tels termes additifs est une possibilité typique de modifier le modèle pour tenir compte de sources d'incertitudes supplémentaires. Le terme additif convient si l'incertitude supplémentaire n'est pas très sensible à la concentration en analyte (ou si la concentration en analyte ne varie pas beaucoup).
Une autre possibilité typique consiste à introduire un terme multiplicatif (ou facteur multiplicatif ). Dans le cas d'une telle modification, l'équation se présenterait comme suit :
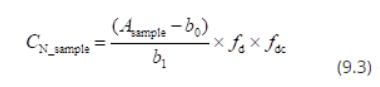
Dans ce cas, la valeur de fdc serait 1 et son incertitude exprimerait l'incertitude relative (c'est-à-dire sans unité) correspondant aux effets possibles de la décomposition et de la contamination. Le terme multiplicatif convient si l'incertitude supplémentaire est à peu près proportionnelle à la concentration en analyte et si la concentration en analyte varie considérablement. Dans le cas des données de cet exemple, la valeur appropriée de u(fdc) serait u(fdc) = u(∆Cdc) / CN_échantillon = 0,019.
***
[1] Commentaire sur l'effet mémoire comme source d'incertitude: l'effet mémoire est problématique tout d'abord dans le cas de l'analyse de traces et ensuite, si l'analyte est spécifiquement sujet à l'adsorption sur des surfaces en verre ou en plastique. Dans ce cas, bien que la concentration d'ammonium dans l'échantillon soit assez faible, il ne s'agit pas encore d'une véritable analyse de traces. L'ion ammonium n'est en aucun cas une espèce fortement adsorbante. Par conséquent, nous pouvons ignorer l'effet de mémoire comme source d'incertitude.
10.4. Valeurs des données d'entrée
https://sisu.ut.ee/measurement/94-step-4-%E2%80%93-finding-values-input-quantities
Trouver les valeurs des données d'entrées
http://www.uttv.ee/naita?id=17640
| We can look now how these uncertainty sources are combinated by our model. This can be seen here. All these uncertainty sources are somehow linked to different input quantities in the model. That in our case we can consider that our model is good and is suitable for this analysis that we intend to carry out. The next step is finding values for the input quantities. Let us start by finding the values for the slope and intercept of the calibration graph. Calibration graph perhaps needs some more explanation. I will give a little bit more information about it. For building a calibration graph, we need to prepare a serie of calibration solutions and in our case there are five calibration solutions. We can denote them by small volumetric flasks. So, each of them has different concentration of ammonium. Let us denote the concentration by C1, C2, C3, C4 and C5. With each of these solutions, we make photometric measurement. We measure the absorbance and we get the absorbance values which we can denote likewise by A1, A2, A3, A4 and A5. In fact, calibration graph is nothing else than the measured quantity which in these case is the absorbance plotted against the concentration. There is the absorbance axis, concentration axis, and these concentrations C1, C2, C3, C4 and C5. It can be seen here. And if we now plot them together with their absorbance values, we'll get a serie of points on this calibration graph. That it will eventually look something like this. Obviously, the absorbance values are here. Good Usually, if the method is linear and spectrophotometry is well known for being highly linear, then the calibration graph can be described by a linear function, meaning a straight line. So here it is. The straight line can always be denoted by a simple linear equation which in our case would be: A is equal to b0 + b1 multiplied by concentration. Those constants b0 and b1 are found from linear regression analysis. There are different factors that cause the uncertainty of this regression coefficient. First of all, all these concentrations, which in turn, have all the volumetric operations as their uncertainty sources and also weighed on standard substance. Secondly, also the uncertainties of the absorbance measurement are also important. So that the uncertainties of all these quantities. We see ten quantities here which in turn have also sub quantities. All these uncertainties finally for us are translated into uncertainties b0 and b1 which eventually take into account the uncertainty due to calibration. Now, if we want to find from this calibration graph the concentration of our sample, then how do we do? We have the absorbance value of the sample. Let's see. And from this absorbance value, we can find the concentration in the sample solution. It's important to note that this C is only the concentration of the sample solution. Not yet the concentration of the analyte in the sample itself. This transformation in mathematical terms would look like this : Or, to be more exact : This is in fact the same as the first term of our model equation that we use for this measurement. An important thing to take into account here is that the absorbance of the sample should be in between the absorbances of the standards meaning that should always be at least one below and at least one above. Then it is correctly carried out. Now this calibration graph can be seen here and the calibration equation is here. With the actual values that we have for our concentrations, and for our absorbances, we get the intercept and slope as can be seen here. So that this equation down here is the actual calibration equation with the real values of slope and intercept uncertainty. Let us now see the other quantities and they have been here now compiled into this table. The absorbance value for sample can be seen here, intercept and slope we spoke about and dilution factor and this additional quantity are seen here. As I already mentioned, this quantity carries value 0 meaning that it does not influence the value of our result. It only influences the uncertainty of our result. |
Nous pouvons maintenant voir comment ces sources d'incertitudes sont combinées par notre modèle. Cela peut être vu ici. Toutes ces sources d'incertitudes sont en quelque sorte liées à différentes quantités d'entrées dans le modèle. Dans notre cas, nous pouvons considérer que notre modèle est bon et convient à l'analyse que nous comptons effectuer. L'étape suivante consiste à trouver les valeurs pour les quantités d'entrées. Commençons par trouver les valeurs de la pente et de l'ordonnée à l'origine de la courbe d'étalonnage. La droite d'étalonnage nécessite peut-être plus d'explications. Je vais donc vous en dire un peu plus. Pour faire une droite d'étalonnage, nous devons préparer une série de solutions d'étalonnages et dans notre cas, il existe 5 solutions d'étalonnages. On peut les représenter par des petites fioles. Ainsi, chacune d'elles a une concentration différente en ammonium. On désigne la concentration par C1, C2, C3, C4 et C5. Avec chacune de ces solutions, nous réalisons des mesures photométriques. Nous mesurons l'absorbance et nous obtenons les valeurs d'absorbances désignées par A1, A2, A3, A4 et A5. En fait, la courbe d'étalonnage n'est rien d'autre que la quantité mesurée qui est dans ce cas l'absorbance tracée en fonction de la concentration. Voici l'axe de l'absorbance, l'axe des concentrations, avec les concentrations C1, C2, C3, C4 et C5 suivantes. On peut le voir ici. Et si nous les associons maintenant avec leurs valeurs d'absorbances, nous obtenons une série de points sur cette droite d'étalonnage. Elle devrait ressembler à quelque chose comme ça. De toute évidence, les valeurs d'absorbances sont ici. Communément, si la méthode est linéaire et que la spectrophotométrie est connue pour être hautement linéaire, alors la courbe d'étalonnage peut être décrite par une fonction linéaire, c'est-à-dire une droite. Alors voilà. La droite peut toujours être désignée par une équation linéaire simple qui dans notre cas serait : A est égal à b0 + b1, multiplié par la concentration. Ces constantes b0 et b1 proviennent de l'analyse de la régression linéaire. Différents facteurs sont à l'origine de l'incertitude de ce coefficient de régression. Tout d'abord, toutes ces concentrations qui, à leur tour, ont toutes les opérations volumétriques comme sources d'incertitudes ainsi que les pesées sur la substance de référence. Ensuite, les incertitudes de mesure sur l'absorbance sont également importantes. Ce sont les incertitudes de ces quantités. Nous voyons ici 10 quantités qui ont à leur tour des sous-quantités. Toutes ces incertitudes se traduisent finalement par les incertitudes b0 et b1 qui prennent éventuellement en compte l'incertitude due à l'étalonnage. Maintenant, si nous voulons trouver à partir de cette droite d'étalonnage la concentration de notre échantillon, comment faisons-nous ? Nous avons la valeur de l'absorbance de l'échantillon. Voyons voir. A partir de cette valeur d'absorbance, nous pouvons retrouver la concentration dans la solution échantillon. Il est important de noter que ce C n'est que la concentration de la solution d'échantillon. Ce n'est pas encore la concentration de l'analyte dans l'échantillon en lui-même. Cette transformation, en termes mathématiques, ressemblerait à ceci, ou pour être plus précis, il s'agit en fait du premier terme de notre modèle d'équation utilisé pour cette mesure. Une chose importante à prendre en compte ici est que l'absorbance de l'échantillon doit être comprise entre les absorbances des étalons ce qui signifie qu'il doit toujours y avoir au moins une valeur en dessous et une au-dessus. Ainsi, il est correctement effectué. Maintenant cette droite d'étalonnage peut être observée ici et l'équation d'étalonnage ici. Avec les valeurs réelles que nous avons pour nos concentrations et pour nos absorbances, nous obtenons l'ordonnée à l'origine et la pente comme on peut le voir ici. Alors que cette équation en bas est l'équation d'étalonnage réelle avec les valeurs réelles des incertitudes de la pente et de l'ordonnée à l'origine. Voyons maintenant les autres quantités et elles sont ici regroupées dans ce tableau. La valeur d'absorbance de l'échantillon est indiquée ici, l'ordonnée à l'origine et la pente dont nous avons parlé ainsi que le facteur de dilution avec cette quantité supplémentaire sont indiqués ici. Comme je l'ai déjà dit, cette quantité porte la valeur de 0 ce qui signifie qu'elle n'influence pas notre résultat. Elle influence seulement l'incertitude de notre résultat. |
Les valeurs de Aéchantillon, b1, b0 et fd sont trouvées à partir des données mesurées (b1 et b0 proviennent de l'analyse de la régression linéaire). La valeur de ΔCdc est nulle.
10.5. Incertitude-type des données d'entrée
https://sisu.ut.ee/measurement/95-step-5-%E2%80%93-standard-uncertainties-input-quantities
Incertitude type des données d'entrée
http://www.uttv.ee/naita?id=17641
| The next step is finding standard uncertainties of the input quantities. We will now look at all the input quantities that we had in our model one by one. And the first input quantity is absorbance of the sample solution. As we saw previously when we looked at uncertainty sources, there are three main uncertainty sources to this input quantity : repeatability of the photometric reading, possible drift of the photometer properties and also possible chemical interferences which can come either by increased absorbance because some other compounds also absorb at used wavelength or by decreased absorbance because something causes the dye either not to form or to a little bit partially decompose. And here the standard uncertainties corresponding to those uncertainty sources are presented. The repeatability standard uncertainty can be easily determined in the laboratory just by measuring repeatedly absorbance of the same solution over a few hours of time. But of course, each time the spectrophotometer cell with the solution should be inserted again and then removed. Now the drift uncertainty can likewise be determined in the laboratory but also it sometimes can be found in the documentation of the spectrophotometer. And what can be said here is that these values correspond to quite safe estimates for contemporary normal usual routine spectrophotometer. Now this uncertainty due to the chemical interferences is more difficult to estimate and actually this estimate can only come by experience from the laboratory unless extensive scientific research is undertaken. In the case of ammonium determination, in most samples we have found that this uncertainty estimate 0.003 absorbance units is fairly useful for the concentration range that is currently under investigation. And now since all these uncertainty components are related to the same quantity, expressed in the units of that same quantity and also expressed as standard uncertainties, we can simply combine them using the squared summing rule which we have seen already previously. We square all the components, sum them up, take square root and find that the combined standard uncertainty of absorbance of the sample solution is 0.0034 absorbance units. The next input quantities are the intercept and slope of the calibration line : b0 and b1. And here we take an approximate solution or an approximate approach. We estimate the standard uncertainties of these two input quantities as simple standard deviations of those quantities as found by regression data analysis, so that their standard uncertainties are given here. And now what does this approximation mean ? It means two things : on one hand it takes into account only the random effects that are evident in these quantities, meaning possible systematic effects will be rejected. However usually such systematic effects and calibration graphs, if they are correctly made, are quite small. Nevertheless, we slightly underestimated uncertainty. On the other hand, all the equations that we are looking at and in particular the equation that comes at the end for calculating the combined standard uncertainty of our measurand they assume non-correlated input quantities. Now, slope and intercept of the same calibration line are always slightly correlated but this correlation is a negative correlation meaning if one of them becomes higher then the other one becomes lower and if a negative correlation is left out of consideration then we slightly overestimate the uncertainty. So, we on one hand somewhat underestimate measurement uncertainty. On the other hand we somewhat overestimate measurement uncertainty. These two effects largely cancel resulting in a very realistic uncertainty estimate. The next input quantity is the dilution factor and dilution factor is nothing else than ratio of different volumes. In our case 50 to 40 ratio resulting in the dilution factor value 1.25. And in this case we will not look in detail at all the volumetric operations because as we will discover later on, the volumetric uncertainty in this uncertainty batch is not very significant. Instead we take a rather safe and very effective approach. We estimate the standard uncertainty of dilution factor as 0.5% of the dilution factor value. As we found according to the experience from our laboratory, this is almost always very suitable uncertainty estimate if volumetric operations are performed correctly. So that the dilution factor standard uncertainty will be 0.5% from 1.25 meaning 0.0063 and it is a unitless quantity. The next input quantity is the ΔCdc. As we saw, this input quantity is meant for taking into account possible decomposition of ammonium or possible contamination of the sample of the solution. I stress here the word possible because it can well be that there's neither contamination nor decomposition but since we do not exactly know, we have to take this possible effect into account as an uncertainty. And this uncertainty unless serious scientific research is carried out, cannot be easily rigorously estimate. So that again we have to use a value which is based on long-term experience. And at this concentration level, it has been found that 0.004 mg/L as standard uncertainty is a very suitable estimate for normal usual conditions of ammonium determination. Now we have found the standard uncertainties of all the input quantities and this slide demonstrates the summary of this. We now have all values and all standard uncertainties and we now can proceed with uncertainty estimation. | L'étape suivante consiste à trouver les incertitudes types des données d'entrée. Nous allons maintenant aborder l'ensemble des données d'entrée que nous avions dans notre modèle une à une. Et la première donnée d'entrée est l'absorbance de la solution d'échantillon. Comme nous l'avons vu précédemment lorsque nous avons abordé les sources d'incertitude, il en existe trois principales qui sont liées à cette donnée d'entrée : la répétabilité de la lecture photométrique, l'éventuelle dérive des propriétés du photomètre et aussi d'éventuelles interférences chimiques qui peuvent provenir soit d'une augmentation de l'absorbance causée par d'autres composés qui absorbent également à la longueur d'onde utilisée ou soit d'une diminution de l'absorbance causée par quelque chose qui empêche la teinte de se former ou qui entraîne en partie sa décomposition. Les incertitudes types qui correspondent à ces sources d'incertitude sont présentées ici. L'incertitude type de répétabilité peut être facilement déterminée au sein du laboratoire en mesurant à plusieurs reprises l'absorbance d'une même solution sur une période de quelques heures. La cellule du spectrophotomètre qui contient la solution doit évidemment être insérée puis retirée lors de chaque mesure. On peut également déterminer l'incertitude de dérive au sein du laboratoire mais on peut aussi parfois la trouver dans la documentation du spectrophotomètre. Ce que l'on peut dire ici c'est que ces valeurs correspondent à des estimations assez certaines pour un spectrophotomètre de routine habituel et récent. L'incertitude causée par les interférences chimiques est plus difficile à estimer et en réalité cette estimation ne peut être trouvée que grâce à l'expérience acquise au sein du laboratoire à moins que des recherches scientifiques approfondies ne soient entreprises. Dans le cas de la détermination de l'ammonium, dans la plupart des échantillons nous avons constaté que cette estimation de l'incertitude qui est à 0,003 unités d'absorbance est assez utile pour la gamme de concentration qui est actuellement étudiée. Puisque toutes les composantes de l'incertitude sont à la fois liées à la même donnée, exprimées en unités de cette même donnée et également exprimées comme incertitudes types, nous pouvons simplement les associer en utilisant la règle de sommation au carré que nous avons vu précédemment. On élève au carré tous les composants, on les additionne, on prend la racine carrée et on trouve que l'incertitude-type composée de l'absorbance de la solution d'échantillon est de 0,0034 unités d'absorbance. Les autres données d'entrées sont l'ordonnée à l'origine et la pente de la droite d'étalonnage: b0 et b1. Ici on prend une solution approximative ou on se place dans une approche approximative. On estime que les incertitudes types de ces deux données d'entrée sont de simples écarts-types de ces données, trouvés par l'analyse des données de régression, de sorte que leurs incertitudes types sont données ici. Que signifie cette approximation? Cela signifie deux choses : d'une part elle ne prend en compte que les effets aléatoires qui sont évidents dans ces données, ainsi les éventuels effets systématiques seront négligés. Cependant, les effets systématiques sont généralement assez faibles si les graphiques d'étalonnage sont correctement réalisés. Néanmoins, nous avons légèrement sous-estimé l'incertitude. D'autre part, les équations que nous examinons et en particulier l'équation qui vient à la fin pour calculer l'incertitude type composéede notre mesurande supposent toutes des données d'entrée non corrélées. La pente et l'ordonnée à l'origine de la même droite d'étalonnage sont toujours légèrement corrélés mais cette corrélation est négative, cela signifie que si l'une augmente alors l'autre diminue et si l'on ne tient pas compte d'une corrélation négative alors on surestime légèrement l'incertitude. D'une part, on sous-estime ainsi quelque peu l'incertitude de mesure. D'autre part on surestime quelque peu l'incertitude de mesure. Ces deux effets s'annulent en grande partie, on obtient ainsi une estimation très réaliste de l'incertitude. La prochaine donnée d'entrée est le facteur de dilution et ce facteur n'est rien d'autre qu'un rapport de différents volumes. Dans notre cas, il s'agit du rapport de 50 sur 40. On obtient un facteur de dilution de 1,25. Dans ce cas nous ne regarderons pas en détail toutes les opérations volumétriques car, comme nous le verrons plus tard, l'incertitude volumétrique dans ce groupe d'incertitudes n'est pas très significative. Au lieu de cela, nous adoptons une approche plutôt sûre et très efficace. On estime que l' incertitude type du facteur de dilution représente 0,5% de la valeur de ce facteur de dilution. Comme nous l'avons constaté lors de l'expérience réalisée dans notre laboratoire, l'estimation de l'incertitude est presque toujours très convenable si les opérations volumétriques sont effectuées correctement. L'incertitude type du facteur de dilution sera ainsi de 0,5% de 1,25 ce qui correspond à 0,0063 et cette donnée obtenue est sans unité. Une autre donnée d'entrée est le ΔCdc. Comme nous l'avons vu, le rôle de cette donnée d'entrée est de prendre en compte la décomposition possible de l'ammonium ou la contamination possible de l'échantillon de la solution. J'insiste ici sur le mot possible car il se peut bien qu'il n'y ait ni contamination ni décomposition mais comme nous ne le savons pas exactement, nous devons tenir compte de cet effet possible comme d'une incertitude. Cette incertitude ne peut pas être facilement évaluée avec rigueur à moins que des recherches scientifiques sérieuses ne soient effectuées. C'est pour cela que nous devons à nouveau utiliser une valeur basée sur une expérience au long terme. A ce niveau de concentration, on considère qu'une incertitude type de 0,004 mg/L est une estimation très convenable pour les conditions normales normales de détermination de l'ammonium. Maintenant, nous avons trouvé les incertitudes types de toutes les données d'entrée comme le résume cette diapositive. Nous avons maintenant toutes les valeurs et toutes les incertitudes types et nous pouvons dès lors procéder à l'estimation de l'incertitude. |
Trouver les incertitudes types des données d’entrée :
L’incertitude type de l’absorbance de l’échantillon (Aéchantillon) est trouvée à partir des 3 composantes de l’incertitude suivantes :
1. L’incertitude liée à la répétabilité de la mesure photométrique :
u (Aéchantillon, rep) = 0.0010 UA (9.2)
Cette incertitude englobe la répétabilité de l’instrument, la répétabilité du positionnement de la cellule de détection, et les possibles interférences, comme des particules de poussières sur les fenêtres optiques de la cellule.
2. L’incertitude liée à l’éventuelle dérive des paramètres du spectrophotomètre :
u (Aéchantillon, dérive) = 0.0012 UA (9.3)
3. L’incertitude liée aux possibles effets d’interférence :
u (Aéchantillon, chim) = 0.0030 UA (9.4)
Elle peut être causée par d’autres composés (interférents) qui absorberaient (ou diffuseraient) la lumière à la longueur d’onde utilisée pour la mesure (menant à l’augmentation de la valeur d’absorbance) ou à des perturbations dans le complexe photométrique en formation (menant à la diminution de la valeur d’absorbance).
En résulte :

Nous voyons que l’incertitude liée à de possibles interférences domine de part sa contribution à l’incertitude totale de l’absorbance de l’échantillon. Bien que dans cet exemple quantifier d’éventuelles interférences est facile, en réalité l’évaluation quantitative de l’incertitude liée aux possibles interférences est difficile. Quelques conseils pratiques à ce sujet sont donnés plus bas dans la section “Évaluer l’incertitude liée aux possibles interférences”.
Les incertitude types de la pente b1 et de l’ordonnée à l’origine b0 sont retrouvées comme des écarts-types des coefficients de régression respectifs (voir les fichiers excel de la section 9.7). C’est une manière approximative de prendre en compte l'incertitude liée à l’analyse par régression linéaire, car :
- On néglige les effets systématiques qui affectent tous les points de la droite de régression.
- On néglige la corrélation négative entre b1 et b0 (qui existe toujours).
Le premier de ces effets mène à une sous-estimation de l’incertitude et le second à une surestimation de l’incertitude. Donc, cette approche ne devrait être utilisée que si on ne s’attend pas à ce que l’analyse par régression linéaire soit parmi les principaux contributeurs à l’incertitude. [1]
L’incertitude liée au facteur de dilution u (fd) est trouvée en émettant l’hypothèse que l’incertitude-type relative composée de toutes les opérations volumétriques concernées ne dépasse pas 0.5%. Si les opérations volumétriques sont menées consciencieusement, alors cette hypothèse est raisonnable dans des conditions usuelles de manipulations en laboratoire. Si nous considérons que la valeur de fd est de 1.25 (sans unités), nous obtenons :
u (fd) = 1.25 x 0.5% / 100% = 0.0065 (sans unités) (9.6)
L’incertitude de ΔCdc compte pour la possible décomposition du complexe photométrique et la possible contamination de l’échantillon. Le mot “possible” est accentué ici : il est bien possible qu’en fait, il n’y ait ni de décomposition du complexe photométrique, ni de contamination de l’échantillon. Cependant, afin de rigoureusement établir ceci, des recherches extensives seraient nécessaires. Par conséquent dans cet exemple, nous utilisons une estimation basée sur l’expérience du laboratoire :
u (ΔCdc) = 0.004 mg/L
Les incertitudes types des quantités d’entrée sont résumées dans le tableau suivant :
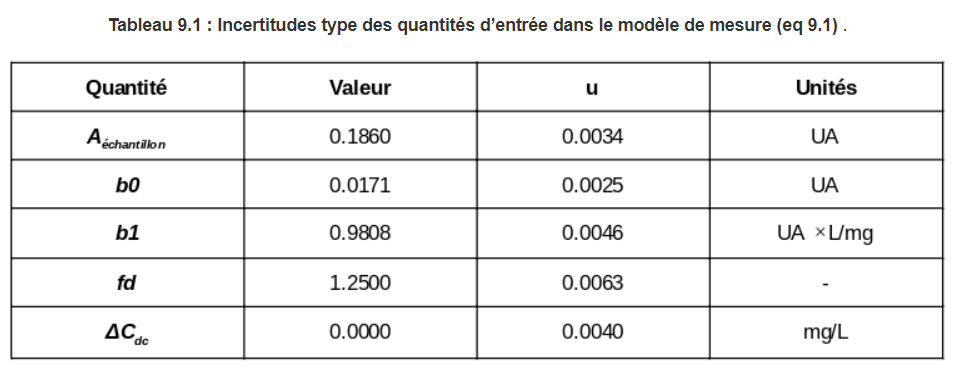
Évaluer l’incertitude liée aux possibles interférences :
Il n’existe pas de manière universellement applicable pour évaluer l’incertitude liée à de possibles interférences. L’approche la plus rigoureuse est de déterminer séparément le contenu des composés interférents dans l’échantillon et de corriger le résultat (alors évidemment l’incertitude de correction doit être évaluée, mais cela est facile). Cependant, dans la plupart des cas, mener des analyses séparées pour déterminer les (possibles) interférents est bien trop coûteux en travail pour être pratique. La chose est d'avantage rendue compliquée par la quantité limitée d’informations généralement disponibles sur les interférents : dans la plupart des cas, on ne sait pas quels sont les composés qui causent l’interférence. Pour cette raison, si l’interférence n’est pas trop importante alors l’approche habituelle est d’essayer de la prendre en compte en augmentant l’incertitude de mesure [2].
Les 2 exemples qui suivent traitent de comment l’incertitude liée à de possibles composés interférents peut être prise en compte dans l’estimation de l’incertitude de mesure.
1- Obtenir et utiliser des informations sur l’interférence à partir du spectre.
Le schéma qui suit présente une situation où l’interférence est clairement présente et peut être observée à partir de l’apparence du spectre :
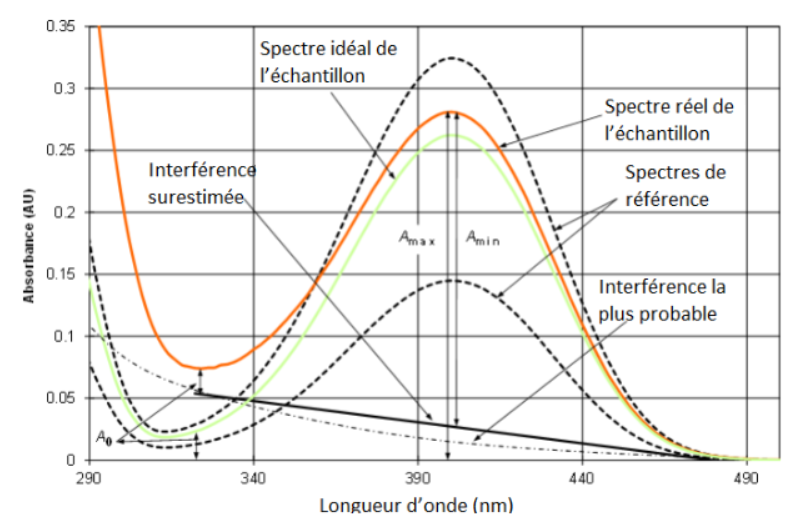
Figure 9.2 : Spectres UV-Visible des solutions de calibration (lignes pointillées), de la solution d'échantillon avec une interférence (rouge), et d'une solution idéale d'échantillon où aucune interférence n'est présente.
Les spectres en pointillés sont ceux de l’analyte dans les solutions de calibration (spectres de référence) où la forme correcte du spectre peut être observée. La ligne verte correspond au spectre “théorique” - c.a.d dépourvu de toute interférence - de la solution d’échantillon. Le spectre le plus probable de l’interférent [3] est présenté par la ligne traitillée. Sa forme suggère que c’est la somme d’un large nombre de différents composés organiques - situation assez commune.
L’absorbance à 400 nm (longueur d’onde de l’absorbance maximum) correspond au signal analytique. Il est évident quand l’on regarde le schéma que si l’on mesure l’absorbance maximale à partir du spectre rouge, on a une absorbance surestimée. Utiliser le spectre vert ou le spectre de l’interférent est impossible, car aucun des deux n’est accessible.
Dans cette situation, on peut essayer d’estimer les valeurs probables d’absorbance maximum et minimum (A) de l’analyte, correspondant respectivement à l’interférence minimale et à la maximale. Une bonne estimation du maximum de A est Amax (cela correspond à la situation très peu probable qu’il n’y a pas d’interférence à 400 nm et que tout le A est dû à l’analyte). Pour estimer le minimum de A on émet l’hypothèse qu’il y a une relation linéaire entre l’effet interférent et la longueur d’onde, comme présenté par la ligne en trait plein nommée “Interférence surestimée”. Elle est surestimée au sens où l’absorbance à 400 nm est plus grande que l’interférence probable. Cette ligne est définie comme suit. Elle est fixée à 0 à 480 nm (il est clair qu’il n’y a pas d’interférence à cette longueur d’onde). A 322 nm la ligne est fixée de manière à ce que la différence d'absorbance du spectre avec l’interférence est la même que la différence du spectre (estimé) sans interférence avec le 0 à la longueur d’onde 322 nm (notée A0 toutes les deux sur le schéma).
A partir des valeurs de Amin et Amax , la valeur d’absorbance corrigée (à utiliser pour le calcul du résultat), ainsi que sa composante de l’incertitude liée à l’interférence peuvent être déterminées comme suit :
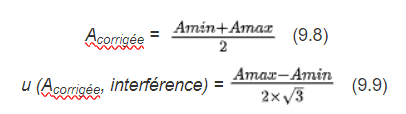
Cette approche est approximative mais son avantage est qu’elle peut être utilisée dans le cas où on ne connaîtrait pas les interférents.
2- Utiliser les données d’interférences de précédentes études.
Si la validation inclut des études d’interférences et que celles-ci sont quantitativement décrites alors ces données peuvent être utilisées pour corriger le résultat et obtenir l’estimation de l’incertitude liée aux possibles interférences. A titre d’exemple, intéressons nous aux valeurs de sélectivité dans la méthode standard ISO 7150:1984 de la détermination spectrophotométrique de la concentration en azote sous forme ammonium. Le tableau 9.2 présente les données concernant l’influence des interférents sélectionnés.
Tableau 9.2 : Influence d’interférents sélectionnés sur la détermination de la concentration en azote sous forme ammonium selon l’ISO 7150:1984 (données tirées de l’ISO 7150:1984).
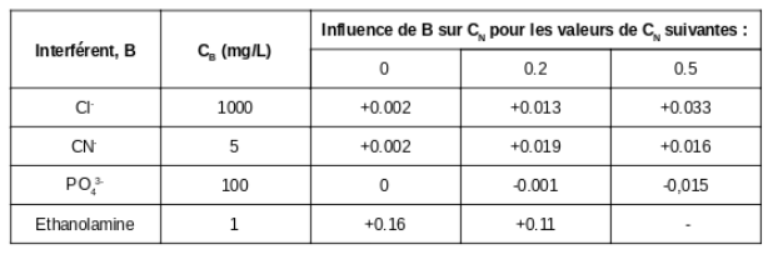
Prenons l’interférence liée au chlore comme exemple. Supposons que nous savons que notre échantillon peut contenir du chlore dans une quantité certainement inférieur à 800 mg/L et que nous avons déterminé une valeur de CN (concentration en azote) d’environ 0.2 dans l’échantillon. Dans ce cas l’interférence maximale serait 0.0013 x 800 / 1000 = 0.0104 mg/L. Si l’on ne connaît pas la quantité de chlore dans l’échantillon et si la déterminer séparément s’avère peu pratique, alors nous pouvons faire l’hypothèse que la concentration en chlore est de (400 ± 400) mg/L. Cette gamme d’incertitude englobe toute la plage de concentrations de 0 à 800 mg/L. L’interférence correspondant à la teneur en chlore serait de +0.0052 mg/L et cette valeur peut être utilisée pour corriger la valeur de CN obtenue (en la soustrayant à CN). Son incertitude type, si on suppose une distribution rectangulaire, est de 0.0054 /  = 0.0030 mg/L. Cette incertitude devrait être incluse additionnellement dans les contributions à l’incertitude.
= 0.0030 mg/L. Cette incertitude devrait être incluse additionnellement dans les contributions à l’incertitude.
3- Utiliser les données sur les interférences via une équation modèle.
Parfois le modèle de la mesure peut directement être utilisé pour prendre en compte les interférences. Un exemple de cette situation peut être retrouvé dans le Test 9.9C.
Autoévaluation sur cette partie du cours : Test 9.5
***
[1] Le même exemple rigoureusement résolu est disponible sur http://www.ut.ee/katsekoda/GUM_examples/. Regardez l’exemple “Ammonium by Photometry” avec le niveau d’élaboration “High (uncertainty estimated at full rigor, suitable for experts)” s’il vous plaît. Comparaison des incertitudes-type composée obtenues : 0,00686 obtenue ici (section 9.7) et 0,0065 mg/L obtenue en toute rigueur. Cela montre que cette approche est acceptable, plus particulièrement car elle mène à une surestimation de l’incertitude plutôt qu’à une sous-estimation de celle-ci.
[2] Si l’interférence est vraiment forte pour une matrice spécifique alors la procédure d’analyse devrait être modifiée et re-validée.
[3] Cette forme du spectre est observée approximativement dans la plupart des cas, si de nombreux composés différents sont présents, chacun à un niveau très faible. De plus, la manière dont le spectre de l’analyte est déformé implique une forme du spectre de l’interférent telle que celle décrite.
10.6. Valeur de la grandeur de sortie
https://sisu.ut.ee/measurement/96-step-6-%E2%80%93-calculating-value-output-quantity
Calculer la valeur de la grandeur de sortie
http://www.uttv.ee/naita?id=17642
| The next step is calculating the measurand value. And as we have the value of all our input quantities, we have the measurand total. Then calculating the measurand value, meaning the ammonium nitrogen concentration is straight forward. The measurand value in this case is 0,215 mg/l. | La prochaine étape est le calcul de la valeur du mesurande. Puisque nous avons toutes les valeurs de quantités d'entrée, nous avons accès au mesurande total. Nous pouvons ensuite calculer la valeur du mesurande, c'est-à-dire la concentration en ammonium. La valeur du mesurande est dans ce cas de 0,215 mg/L. |
La valeur de la grandeur de sortie (la valeur du mesurande) est calculée à partir des valeurs de quantité d'entrée (section 9.4) en utilisant le modèle mathématique (section 9.2). La valeur du mesurande dans cet exemple est la suivante : CN_échantillon = 0.215 mg/L.
10.7. Incertitude-type composée
https://sisu.ut.ee/measurement/97-step-7-%E2%80%93-calculating-combined-standard-uncertainty
Calculer l'incertitude type composée
http://www.uttv.ee/naita?id=17643
| The next step is finding the combined standard uncertainty of the output quantity and if we have non correlating input quantities then the following equation holds as we have seen already earlier. And all these terms here that are squared in this equation are called uncertainty components so that for each and every of our input quantities we have one uncertainty component. And if we now put our concrete data into this equation then we arrive at the following and this already straightforwardly corresponds to our measurement situation. If we look at it we see that each uncertainty component in turn is composed of two parts, one is the standard uncertainty of the specific input quantity expressed in the units of that same input quantity and the other one is the partial derivative of the output quantity with respect to that input quantity. It's actually the partial derivative that converts all the input quantity units into the output quantity units so that we are allowed to sum up all these terms. And also the partial derivative sometimes is called sensitivity coefficient because it shows how sensitivity uncertainty of the output quantity is towards the uncertainty of the input quantity. This equation may look problematic to calculate all these derivatives and so on but we see in a separate demonstration that it can be easily calculated using a spreadsheet software according to the crowd and approach. So that we will be able to calculate easily with the software directly these uncertainty components and on this slide we already have them calculated and they are presented here. So all the uncertainty components of input quantities are listed here. We see that some of them are positive and some are negative. This is fully normal. Those quantities which on increasing of their value also increase the value of the output quantity have positive signs but the rules which one increase decrease the output quantity value have a negative sign. It is also important to stress as I said already previously that strictly changing this equation works for non correlating input quantity as we saw in the previous lecture in fact slope and intercept, two of our input quantities, are correlated but as I explained there this correlation in this particular case is not the problem so that it will be small and it actually constants another effect so that we do not really need to worry about correlation in this particular case. And if we now combine all the uncertainty components according to this equation we find the combined standard uncertainty of the zone 0.00686 mg/L. | La prochaine étape est de trouver l’incertitude type composée de la quantités de sortie et si nous avons des quantités d’entrée non corrélées, alors l’équation suivante fonctionne comme nous l’avons déjà vu précédemment. Et tous ces termes ici, qui sont au carré dans cette équation, sont appelés les composantes d’incertitudes et donc pour chacune et pour toutes nos quantités d’entrée nous avons une composante d’incertitude. Et si nous rentrons maintenant nos données concrètes dans cette équation alors nous obtenons ceci et cela correspond directement à notre situation de mesure. Si nous regardons bien nous voyons que chaque composante d’incertitude à son tour est composée de deux parties, une est l’incertitude type de la quantité d’entrée spécifique exprimée dans l’unité de cette même quantité d’entrée et l’autre est la dérivée partielle de la quantité d’entrée en respectant la quantité d’entrée. C’est en réalité la dérivée partielle qui convertit toutes les quantités d’entrée en unité de quantité de sortie afin que nous soyons autorisés à résumer tous ces termes. Et également la dérivée partielle est parfois appelée coefficient de sensibilité parce qu’elle montre la sensibilité de l’incertitude sur la quantité de sortie vers la sensibilité de la quantité d’entrée. Cette équation semble problématique pour calculer toutes ces dérivées et ainsi de suite mais nous voyons dans une démonstration séparée que cela peut être simplement calculé en utilisant un tableur selon l’approche de Kragten. Nous serons donc capables de calculer, facilement avec le tableur, directement ces composantes d’incertitude et sur cette diapositive nous les avons déjà calculées et elles sont présentées ici. Donc toutes les composantes d’incertitude de cette quantité d’entrée sont listées ici. Nous voyons que certaines sont positives et certaines sont négatives. C’est complètement normal. Ces quantités, qui en augmentant leur valeur augmentent également les valeurs de quantité de sortie, ont des signes positifs mais les règles sont que quand une augmente et entraîne une diminution de la quantité de sortie alors elle a un signe négatif. Il est également important de souligner, comme je l’ai déjà dit précédemment, que de changer strictement cette équation fonctionne pour des quantités d’entrée non corrélées comme nous l’avons vu dans la leçon précédente, en réalité la pente et l’interception, deux de nos quantités d’entrée, sont corrélées mais comme je l’ai expliqué ici, cette corrélation dans ce cas particulier n’est pas un problème donc il sera petit et en réalité il garde constant un autre effet afin que nous n’ayons pas vraiment besoin de s’inquiéter à propos de la corrélation dans ce cas particulier. Et si maintenant nous combinons toutes les composantes d’incertitude en s’appuyant sur cette équation nous trouvons l’incertitude type composée à la zone 0.00686 mg/L. |
Calcul numérique des éléments d'incertitudes : la méthode Kragten
http://www.uttv.ee/naita?id=17721
|
I mentioned that these uncertainty components can be calculated numerically and now we will be looking how exactly we do that. And for this we will use the so-called Kragten spreadsheet approach. So that each of these components, which in our case actually look like this, they will be calculated separately so that finally for each of these components we will get numerical values exactly as seen here. And in addition these components are used for calculating the uncertainty contributions or the so-called uncertainty indexes that we saw in the previous lecture. And the uncertainty index of an input quantity x1 is found by dividing its squared uncertainty component with the sum of squares of all the uncertainty components. So that once we have calculated the uncertainty component values using the Kragten approach finding the uncertainty indexes is very easy. Our aim now is to calculate numerically the uncertainty component for our uncertainty estimation whereby the most important thing is numerically calculating the partial derivative. And let us start by writing out the function. Our output quantity is calculated by some function from a number of input quantities and different input quantities. And let us examine for example the uncertainty component corresponding to the input quantity number one, all others go just the same way. So the uncertainty component reads like this. The uncertainty over the output quantity that is caused by this particular quantity. And this is calculated like this. And whenever we calculate numerically partial derivatives we replace these infinitesimally small changes or infinitesimally small Deltas by finite Deltas. So that the essence of approximation that is made is this case is the following. Now with Delta x1 all is here but how would you will find Delta y ? For this we have to look at this function again and it is obvious that we have to substract from the function found at position x1 + Delta x1, the function as it stands here whereby x1 has not been modified. So Delta Y is calculated as follows. So that we substract just like this. And now we can substitute this Delta y here so that the uncertainty component calculation will read as follows. So and now an important thing in numerical calculation of partial derivatives is : how do we choose which value of Delta we use here? And what Kragten proposed, and this is why this method carries his name, he proposed that let us take the Delta equal to the standard uncertainty of this input quantity. So that we can use like this. And the beauty of his proposal is that these two now cancel out. So that eventually the uncertainty component is calculated as follows. And this is the way the Kragten approach works. And we will now illustrate this by calculating as a concrete example the result, the uncertainty of our particular ammonium determination exam. Let us see now how the Kragten approach of calculating the uncertainty components by the numerical method works in practice. I have prepared here an Excel spreadsheet in such a way that we have here all our input quantities. Their values their standard uncertainties and their units and also I have added here the model equation and the equation for calculated the combined standard uncertainty from the uncertainty components. And the first step of setting up a Kragten calculation is building a Kragten matrix. Kragten matrix has as many columns and as many rows as many input quantities we have, meaning in our case it’s a 5 to 5 matrix. And it is useful to specifically mark the diagonal elements of the Kragten matrix as it will be evident later on. This will be used for the calculation in an important way. And now each of the columns in the Kragtend matrix corresponds to one of the input quantities. Therefore it is useful to mark them and this can be done with “copy” and with the so-called “paste special” function whereby we use the “transpose” option so that all the quantity names that initially were in a column are now in a row. The next thing we now have to organize all these quantity values into these columns here. And for doing that we make this one equal to the first value and it is useful now to fix the column D but not the row 7. And this enables us to copy this value throughout this matrix. So we copy now and again we don’t juste paste but to the “paste special” and we paste formulas. So that we see now that in each of the cells here we basically have the same quantities that in the corresponding cell in this column here. And now the diagonal elements come into play. To each of these diagonal values we will now add the standard uncertainty of the respective input quantity. So that here we add the standard uncertainty of a sample. Here we add the standard uncertainty of b0. Here we add the standard uncertainty of b1. Here we add the standard uncertainty of fd. And here we add the standard uncertainty of ΔCdc. So we see now that even we don’t know the values in these columns are still fairly similar. In each column the diagonal value is different from the others. And now we will calculate here and also here the output quantity value and let’s see how that will work. CN sample that does also use the formatting such a way as in towards in other places. Okay and now the value we will find as follows : from Asample we substract b0 we then divide by b1 multiply by the dilution factor and add ΔCdc. Even though its value is 0 it is extremely important to add it because as we see later on in one of the case its value will not be 0 anymore. So and this value of course carries the units milligrams per liter. And now with this same formula which we can copy we can calculate all these values also here. And now we will calculate the uncertainty components as simple substraction between these values that we have here and this value here. Basically all these values are the same as was shown on the whiteboard. The value of the function whereby the one of the input quantities, the Delta input quantity, which according to the Kragten approximation is approximated by standard uncertainty, is added. So components come here and again we substract and we again fix the column. And it is $d13 that we substract. So and if we now copy this component here we can easily find the values of all the uncertainty components. Now it is necessary to calculate the squared components because all of them have to be taken to the square. The component squared we find like this. And it is of course needed now to calculate the sum of the squared components. And this sum is here. And the square root of this sum actually is the combined standard uncertainty of the output quantity and we can calculate it here in this cell. Equal to square root of this. So that it is exactly 0.00686 as we seen in the slides. Based on this data that we have now we also can calculate easily the uncertainty indexes or the uncertainty contributions. And for each input quantity the uncertainty index is calculated by dividing the squared uncertainty component by the sum of squares of all the components. And again it is useful to fix here the column so that we can easily copy them. And these indexes can be formatted as percentages even though at the moment they are expressed simply as ratios. And expressing as percentages we do by formatting cell. As percentage. So these are here, the uncertainty indexes that we saw also in the lecture 9. And we can mark this result in green. So we now have our measurement result and it’s combined standard uncertainty here. And we now can express our measurement result in full using the expanded uncertainty. Let us do that here. Our result is Ammonium nitrogen concentration in the sample. And we start parentheses. The measured value is here. Here we put the plus/minus symbol. And here we put the expanded uncertainty which we in this case express as k = 2 uncertainty. And here the parentheses end. mg/L is the unit. And in our case the coverage factor is equal to two. Importantly we also need to take care of the decimal places. In this case is our first significant digit is 1 so that we express the uncertainty in two significant digits which means three decimal places or three places after the comma. So, and this could be our measurement result. And because, as we saw, we have the reason to expect that in this case the result really does correspond to the normal distribution we also can write here norm. |
J’ai mentionné que ces composantes d’incertitude peuvent être calculées numériquement et maintenant nous allons regarder exactement comment nous faisons ça. Et pour cela nous utiliserons l’approche dite du tableur de Kragten. De cette façon chacune de ces composantes, qui dans notre cas ressemblent à ça, seront calculées séparément de sorte que finalement pour chacune de ces composantes nous allons avoir une valeur numérique, précisément comme vu ici. Et en addition, ces composantes sont utilisées pour calculer les contributions d’incertitude ou aussi appelées les indices d’incertitude que nous avons vu dans le cours précédent. Et l’indice d’incertitude d’une quantité d’entrée x1 est trouvé en divisant son composant d’incertitude au carré par la somme des carrés de toutes les composantes d’incertitude. Donc une fois que nous avons calculé les valeurs de la composante d’incertitude, en utilisant l’approche de Kragten, trouver les indices d’incertitude est très simple. Notre but est maintenant de calculer numériquement la composante d’incertitude pour notre estimation d’incertitude par lequel la chose la plus importante est de calculer numériquement la dérivée partielle. Et nous allons commencer par écrire la fonction. Notre quantité de sortie est calculée par une certaine fonction depuis un nombre de quantités d’entrée et différentes quantités d’entrée. Examinons par exemple la composante d’incertitude correspondant à la quantité d’entrée numéro une, toutes les autres vont de la même façon. Donc la composante d’incertitude est lue de cette façon. L’incertitude sur la quantité de sortie qui est causée par cette quantité en particulier. Et c’est calculé comme ça. Et chaque fois que nous calculons numériquement les dérivées partielles nous remplaçons ces infiniment petits changements ou ces infiniment petits deltas par des deltas finis. De cette façon l’essence de l’approximation qui est faite dans ce cas est la suivante. Maintenant, avec delta x1 tout est ici mais comment vous allez trouver le delta y ? Pour cela nous devons regarder cette fonction encore et il est évident que nous devons soustraire à cette fonction trouvée à la position x1 + delta x1, par la fonction comme elle est ici, où x1 n’a pas été modifié. Donc delta y est calculé comme suit. Comme nous avons juste à soustraire de la sorte. Et maintenant nous pouvons remplacer ce delta y ici pour que le calcul de la composante d’incertitude se lise comme suit. Maintenant une chose importante dans le calcul numérique des dérivées partielles est : comment nous choisissons quelle valeur de delta nous utilisons ici ? Et ce que Kragten propose, et c’est pourquoi cette méthode porte son nom, il propose que nous prenions un delta égal à l’incertitude type de cette quantité d’entrée. De cette façon nous pouvons l’utiliser comme ça. Et la beauté de sa proposition est que ces deux-là maintenant s’annulent. Donc la composante d’incertitude est éventuellement calculée comme suit. Et c’est ainsi que l’approche de Kragten fonctionne. Et maintenant nous allons illustrer ceci en calculant comme un exemple concret le résultat, l’incertitude de notre propre examen sur la détermination de l’ammonium. Nous allons maintenant regarder comment l’approche de Kragten pour calculer les composantes d’incertitude par la méthode numérique fonctionne en pratique. J’ai préparé ici un tableur Excel de sorte à ce que nous ayons ici toutes nos quantités d’entrée. Leur valeur, leur incertitude type et leur unité, et aussi j’ai ajouté ici l’équation modèle et l’équation pour calculer l’incertitude type composée à partir des composantes d’incertitude. Et la première étape pour mettre en place le calcul de Kragten est de construire une matrice de Kragten. Une matrice de Kragten possède autant de colonnes et autant de lignes qu’il y a de quantités d’entrée que nous avons, ce qui veut dire dans notre cas une matrice de 5 par 5. Et il est utile de marquer spécifiquement les éléments en diagonale de la matrice de Kragten et cela sera évident plus tard. Ce sera utilisé pour le calcul d’une façon importante. Et maintenant chacune des colonnes dans la matrice de Kragten correspond à une des quantités d’entrée. Par conséquent, il est utile de les marquer et cela peut être fait en utilisant « copier » et avec la fonction appelée « collage spécial » où nous utilisons l’option « transposer » pour que tous les noms des quantités qui étaient initialement dans une colonne soient maintenant dans une ligne. La prochaine chose que nous devons maintenant faire est d’organiser toutes ces valeurs de quantité dans ces colonnes ici. Et pour faire ça nous mettons celle-ci égale à la première valeur et il est utile maintenant de fixer la colonne D mais pas la ligne 7. Et cela nous empêche de copier cette valeur tout au long de cette matrice. Donc maintenant nous copions et encore une fois nous ne collons pas juste mais nous allons dans « collage spécial » et nous copions « formules ». Donc maintenant nous voyons que dans chacune des cellules nous avons basiquement les mêmes quantités que dans les cellules correspondantes dans la colonne ici. Et maintenant les éléments en diagonale viennent jouer un rôle. Pour chacune de ces valeurs en diagonale nous allons maintenant ajouter l’incertitude type de leur quantité d’entrée respective. Donc ici nous ajoutons l’incertitude type d’un échantillon. Ici nous ajoutons l’incertitude type de b0. Ici nous ajoutons l’incertitude type de b1. Ici nous ajoutons l’incertitude type de fd. Et ici nous ajoutons l’incertitude type de ΔCdc. Donc nous voyons maintenant que même si nous ne connaissons pas les valeurs dans ces colonnes, elles sont toutes très similaires. Dans chaque colonne la valeur en diagonale est différente des autres. Et maintenant nous allons calculer ici et aussi ici la valeur de la quantité de sortie et voyons comment cela fonctionne. CN sample qui utilise aussi le formatage de telle sorte comme suit dans d’autres endroits. Okay et maintenant nous allons trouver la valeur comme suit : Asample on soustrait b0, nous divisons ensuite par b1 et multiplions par le facteur de dilution et on ajoute ΔCdc. Même si cette valeur est 0 il est extrêmement important de l’ajouter car comme nous le verrons plus tard dans un des cas cette valeur ne sera plus 0. Et cette valeur porte bien sûr l’unité mg/L. Et maintenant avec cette même formule, que nous pouvons copier, nous pouvons calculer toutes les valeurs ici. Et maintenant nous allons calculer les composantes d’incertitude comme une simple soustraction entre ces valeurs que nous avons ici et ces valeurs ici. Basiquement, toutes ces valeurs sont les mêmes comme montré sur le tableau. La valeur de la fonction par laquelle l’une des quantités d’entrée, la quantité d’entrée delta, qui selon l’approximation de Kragten est approximée par l’incertitude type, est ajoutée. Donc les composantes viennent ici et encore une fois nous soustrayons et nous fixons encore la colonne. Et c’est calculé comme ça. Et $d13 que nous soustrayons. Et donc si nous copions maintenant cette composante ici nous pouvons facilement trouver les valeurs de toutes les composantes d’incertitude. Maintenant il est nécessaire de calculer les carrés des composantes car il faut tous les mettre au carré. La composante au carré que nous trouvons comme ça. Et il est bien sûr nécessaire maintenant de calculer la somme des carrés des composantes. Et cette somme est ici. Et la racine carrée de cette somme est en réalité l’incertitude type composée de la quantité de sortie et nous pouvons la calculer ici dans cette cellule. Egale à la racine carrée de ça. Donc c’est exactement 0.00686 comme nous l’avons vu dans les diapositives. En se basant sur cette donnée que nous avons maintenant, nous pouvons également calculer facilement les indices d’incertitude ou les contributions d’incertitude. Et pour chaque quantité d’entrée l’indice d’incertitude est calculé en divisant le carré de la composante d’incertitude par la somme des carrés de toutes les composantes. Et c’est encore utile de fixer ici la colonne pour que nous puissions facilement les copier. Et ces indices peuvent être formatés comme des pourcentages même si tout de suite ils sont exprimés simplement comme des ratios. Et exprimer comme des pourcentages nous la faisons en formatant la cellule. Comme pourcentage. Donc ceci est ici, les indices d’incertitude que nous avons aussi vus dans le cours 9. Et nous pouvons marquer ce résultat en vert. Donc maintenant nous avons notre résultat de mesure et c’est une incertitude type composée ici. Et nous pouvons maintenant exprimer notre résultat de mesure en entier en utilisant l’incertitude élargie. Faisons le ici. Notre résultat de la concentration en ammonium dans l’échantillon. Et nous ouvrons les parenthèses. La valeur mesurée est ici. Ici nous mettons le symbole plus/moins. Et ici nous mettons l’incertitude élargie qui dans ce cas est exprimée avec k = 2 fois l’incertitude. Et ici les parenthèses se ferment mg/L est l’unité. Et dans notre cas le facteur de recouvrement est égal à deux. Surtout nous devons aussi faire attention aux décimales. Dans ce cas notre premier chiffre significatif est 1 donc nous exprimons notre incertitude avec deux chiffres significatifs ce qui veut dire qu’il y a trois décimales ou trois places après la virgule. Donc, et ceci pourrait être notre résultat de mesure. Et parce que, comme nous l’avons vu, nous avons une raison de s’attendre dans ce cas à ce que ce résultat corresponde réellement à la distribution normale, nous pouvons aussi écrire ici “norm”. |
Le fichier XLS initial (ne contenant que les données mais pas les calculs) utilisé dans cet exemple et le fichier XLS contenant également le calcul de l'incertitude-type composée (et incertitude élargie) selon l'approche de Kragten peuvent être téléchargés ci-dessous :
uncertainty_of_photometric_nh4_determination_kragten_initial.xls (49KB)
uncertainty_of_photometric_nh4_determination_kragten_solved.xls (53 KB)
10.8. Incertitude élargie
https://sisu.ut.ee/measurement/98-step-8-%E2%80%93-finding-expanded-uncertainty
L'incertitude élargie peut être déterminée avec deux niveaux de sophistications différents. L'approche la plus simple utilise simplement une valeur de k prédéfinie (le plus souvent 2) et la probabilité de recouvrement réelle n'est pas discutée. Cette approche est présentée dans la première conférence vidéo.
Trouver l'incertitude élargie (approche simple)
http://www.uttv.ee/naita?id=17644
|
The next step is finding the expanded uncertainty of the output quantity and the expanded uncertainty is found by multiplying the combine standard uncertainty with a coverage factor. Coverage factor value can be chosen by the person who presents the result but value 2 is very often used. And if the distribution of the output quantity is roughly normal then this uncertainty corresponds to roughly 95% probability. And in our case the expanded uncertainty is 0.014 mg/L. And now we can also present our measurement result: 0.215 plus minus 0.014 mg/L at k 2 level, meaning coverage factor 2 level.
|
La prochaine étape est de trouver l’incertitude élargie de la quantité totale, et l’incertitude élargie est trouvée en multipliant l’incertitude type composée par le facteur d’élargissement. La valeur du facteur d’élargissement peut être choisie par la personne qui présente les résultats, mais la valeur 2 est utilisée dans la plupart des cas. Si la quantité totale suit une loi à peu près normale alors on obtient une incertitude avec une confiance d’environ 95%. Dans notre cas, l’incertitude élargie est de 0,014 mg/L. Nous pouvons donc maintenant présenter le résultat de notre mesure : 0,215 plus ou moins 0,014 mg/L à un niveau k=2, ce qui correspond à un facteur d’élargissement de 2.
|
La deuxième approche est plus sophistiquée. Il s'agit d'une approche d'approximation basée sur l'hypothèse que la fonction de distribution de la variable étudiée peut être approchée par une distribution de Student avec le nombre effectif de degrés de liberté trouvé par la méthode dite de Welch-Satterthwaite. Cela permet ensuite d'utiliser le coefficient de Student correspondant à un niveau de confiance souhaité (probabilité de recouvrement) comme facteur de recouvrement. Cette approche est expliquée dans la deuxième conférence vidéo.
Trouver l'incertitude élargie (la méthode Welch-Satterthwaite)
http://www.uttv.ee/naita?id=17916
|
Let us see now how the Welch-Satterthwaite approach for calculating the effective number of degrees of freedom works in practice. Using the effective number of degrees of freedom we can define the coverage factor which corresponds to a predefined probability. And we will look at this on the example of the ammonium nitrogen uncertainty calculation which was already previously. We calculated there the uncertainty indexes for uncertainty contributions and we saw that there are three quantities that dominate the uncertainty, which enabled us assuming that our output quantity is normally distributed. The Welch-Satterthwaite approach actually also enables verifying how when is good. I have put here the Welch-Satterthwaite equation and the indexes of the input quantities that we need here. We already have. And now we need to calculate de number of degrees of freedom for all of these input quantities. So “df” And let us now look, quantity by quantity. If we look at Asample then Asample had three uncertainty components, the most influential of them is the uncertainty due to the chemical interference and this is a B type uncertainty estimate. And whenever we have B type uncertainty estimate we can say that the number of degrees of freedom is either infinity or at least very large. And very often people use something like 100 or 50 for number of degrees of freedom of such quantities. So we will assume that the number of degrees of freedom of a sample is 50. Now b0 and b1 (the incercept and the slope of the regression line) come from this regression data. We have here five data points. But in linear regression, the number of degrees of freedom is found from the number of data points by subtracting the number of found parameters. And in our case there are two parameters: the slope and the incercept. 5-2 is 3 so in this case we have three uncertainty components both for the slope and for the intercept. Now, dilution factor. It’s uncertainty estimate was a typical B type uncertainty estimate which again enables us saying that the number of degrees of freedom is very large. Let’s say roughly 50 and the same goes about ΔCdc which also is a B type estimated quantity. So number of degrees of freedom is 50. Now we have to calculate the ratios of indexe squared versus the number of degrees of freedom, and this we will do here in this row. Indexe squared divided by 50. Ok so now we have all the ratios here and now the number of effective degrees of freedom for our output quantity. We can calculate in this cell, it is 1 divided by sum of all these ratios and we find that the number of effective degrees of freedom in this case is 45. And in order to find now a coverage factor corresponding to 95% probability with 45 degrees of freedom, we calculate the so-called student coefficient or “t” coefficient. So it is our k value at 95% probability. And this is found by the so-called “tinv” function which uses as its parameters the probability which is expressed as 100 minus the desired probability meaning 5% but this 5% has to be presented as a ratio meaning not 5 but 0.05. And the number of degrees of freedom of course comes from this cell. And we see that the coverage factor corresponding to 95% probability is very similar to its actual 2.14, and this means that we are fully entitled to calculate uncertainty at k2 level and assume the normal distribution. But now, here we can insert the correct rigorously calculated k value, and we see that the uncertainty remains 0.014 just as we found previously with using k2.
|
Voyons maintenant comment l’approche Welch-Satterthwaite pour le calcul du nombre effectif de degrés de liberté fonctionne dans la pratique. En utilisant le nombre effectif de degrés de liberté nous pouvons définir le facteur de recouvrement qui correspond à une probabilité prédéfinie. Et nous allons examiner cela avec l’exemple du calcul d’incertitude sur l’azote ammoniacal que l’on a déjà fait auparavant. Nous avions calculé les indices d’incertitude pour les contributions à l’incertitude et nous avions vu qu’il y a trois quantités qui prédominent dans l’incertitude, ce qui nous a permis de supposer que notre quantité de sortie est normalement distribuée. L’approche Welch-Satterthwaite permet également de vérifier comment elle se maintient. J’ai mis ici l’équation de Welch-Satterthwaite et les indices des quantités d’entrée dont nous avons besoin ici. Que nous avons déjà. Et maintenant nous devons calculer les nombres de degrés de liberté pour toutes ces quantités d’entrée. Donc « df » Et regardons maintenant, quantité par quantité. Si nous regardons Asample (Aéchantillon) alors Asample (Aéchantillon) avait trois composantes d’incertitude, la plus influente d’entre elles est l’incertitude due aux interférences chimiques et il s’agit d’une incertitude estimée de type B. Et chaque fois que nous avons une estimation d’incertitude de type B, nous pouvons dire que le nombre de degrés de liberté est soit infini, soit au moins très grand. Et très souvent les gens utilisent quelque chose comme 100 ou 50 pour le nombre de degrés de liberté de ces quantités. Nous supposerons donc que le nombre de degrés de liberté d’un échantillon est de 50. Maintenant b0 et b1 (l’ordonnée à l’origine et la pente de la régression linéaire) proviennent des données de cette régression. Nous avons ici cinq points de données. Mais en régression linéaire, le nombre de degrés de liberté est trouvé à partir du nombre de points de données en soustrayant le nombre de paramètres trouvés. Et dans notre cas, il y a deux paramètres : la pente et l’ordonnée à l’origine. 5-2 fait 3, donc dans ce cas nous avons trois composantes de l’incertitude, tant pour la pente que pour l’ordonnée à l’origine. Maintenant, le facteur de dilution. Son incertitude estimée était typiquement une incertitude estimée de type B, qui encore une fois nous permet de dire que le nombre de degrés de liberté est très grand. Disons environ 50 et la même chose pour ΔCdc qui est aussi une quantité estimée de type B. Donc le nombre de degrés de liberté est de 50. Maintenant nous devons calculer les ratios des indices au carré en fonction du nombre de degrés de liberté, et c’est ce que nous allons faire ici dans cette ligne. Indice au carré divisé par 50. Ok, donc maintenant nous avons tous les ratios ici et maintenant le nombre effectif de degrés de liberté pour notre quantité de sortie. Nous pouvons le calculer dans cette cellule, c’est 1 divisé par la somme de tous ces ratios et nous constatons que le nombre de degrés de liberté dans ce cas est de 45. Et afin de trouver maintenant un facteur de recouvrement correspondant à une probabilité de 95% avec 45 degrés de liberté, nous calculons le coefficient dit « de Student » ou coefficient « t ». Il s’agit donc de notre valeur de k à une probabilité de 95%. Et nous le trouvons grâce à la fonction « tinv » qui utilise comme paramètres la probabilité qui est exprimée comme 100 moins la probabilité souhaitée c'est-à-dire 5%, mais ces 5% doivent être présentés comme un ratio c'est-à-dire non pas 5 mais 0,05. Et le nombre de degrés de liberté vient bien sûr de cette cellule. Et nous voyons que le facteur de recouvrement correspondant à une probabilité de 95% est très similaire à 2 : il est enfait de 2.14, et cela signifie que nous avons pleinement le droit de calculer l’incertitude au niveau k2 et de supposer la distribution normale. Mais maintenant, nous pouvons insérer ici la valeur correcte de k, rigoureusement calculée, et nous voyons que l’incertitude reste de 0,014 juste comme nous l’avions constaté précédemment avec l’utilisation de k2.
|
Le fichier Excel contenant l'incertitude-type composée et le calcul de l'incertitude élargie et le fichier Excel contenant le calcul de l'incertitude élargie à l'aide du facteur de couverture trouvé en utilisant le nombre effectif de degrés de liberté de l'approche Welch-Satterthwaite peuvent être téléchargés ici :
uncertainty_of_photometric_nh4_determination_kragten_solved.xls
uncertainty_of_photometric_nh4_determination_kragten_solved_df.xls
10.9. Analyse de l'incertitude obtenue
https://sisu.ut.ee/measurement/99-step-9-%E2%80%93-analysis-uncertainty-sources-and-conclusions
Le calcul des composantes de l'incertitude (indices d'incertitude) est expliqué et démontré au point 9.7.
Analyse des sources d'incertitudes
http://www.uttv.ee/naita?id=17645
|
The next tip is analysing the uncertainty contributions. The uncertainty contributions can be calculated, meaning it is possible to calculate how many % of the overall uncertainty is caused by any of these uncertainty contributions. And we see that in our case, first of all, the largest share of uncertainty, almost 40%, comes from the absorbance measurement of the sample. Secondly, almost equal part comes from ΔCdc, meaning from the possible decomposition of contamination. The next is the regression analysis and it is useful to look at it as one uncertainty contribution of 24% roughly. And the dilution factor meaning the volumetry cooperations made with the sample solution count for only 3% of the overall uncertainty. These 3% justifies why we were able to use the simplified approach of accounting for volumetric uncertainty. Now, an important thing here is that we have 3 uncertainty sources of comparable magnitude. And as I explained in one of the previous lecture, if these holds if there are at least 3 uncertainty sources of comparable magnitude, then we can assume that our output quantity indeed is normally distributed. So that in this particular case, we are fully allowed to write the word norm here, meaning we are allowed to assume that this output quantity really is distributed according to the normal distribution. These uncertainty contributions are also very useful in looking at how can we optimize our measurement procedure or how can we further decrease the uncertainty. Obviously we should put effort into the input quantities which have the largest share of the uncertainty so that the absorbance measurement of the sample and the possible contaminational decomposition is something to take care of. So that every effort should be devoted to possible spectral interference, possible instability of the dye, possible contamination, possible decomposition. However all in all, what can be said about this procedure, is as it already can be considered fairly optimized. And how do we see that again if there are several uncertainty sources, that are of comparable magnitude, this means that the procedure is quite optimize. If there was just 1 single uncertainty source with, let’s see, 85% contribution then we could say that yes this particular input quantity needs lot of attention and we cannot call our measurement procedure, optimized. |
L’astuce suivante est d’analyser les contributions de l’incertitude. Les contributions de l’incertitude peuvent être calculées, ce qui signifie qu’il est possible de calculer combien de % de l’incertitude globale est causé par l’une de ces contributions de l’incertitude. Et nous voyons que dans notre cas, premièrement, la plus grande part d’incertitude, environ 40%, vient de la mesure d’absorbance de l’échantillon. Deuxièmement, une part presque égale provient de ΔCdc, ce qui veut dire la décomposition possible de la contamination. La suivante est l’analyse de régression et cela est utile de la considérer comme une contribution de l’incertitude de 24% environ. Et le facteur de dilution signifiant les coopérations volumétriques réalisées avec la solution échantillon ne comptent que pour 3% seulement de l’incertitude globale. Ces 3% justifient pourquoi nous sommes capables d’utiliser l’approche simplifiée pour tenir compte de l’incertitude volumétrique. Maintenant, une chose importante ici est que nous avons 3 sources d’incertitudes d’ampleur comparable. Et comme je l’ai expliqué dans l’une des leçons précédentes, si cela se produit s’il y a au moins 3 sources d’incertitudes d’amplitude comparable, alors nous pouvons supposer que notre quantité de sortie est en effet normalement distribuée. De sorte que dans ce cas particulier, nous sommes pleinement autorisés à écrire le mot norme ici, ce qui signifie que nous sommes autorisés à supposer que cette quantité de sortie est vraiment distribuée selon la distribution normale. Ces contributions de l’incertitude sont également très utiles en regardant comment nous pouvons optimiser notre procédure de mesures ou comment pouvons-nous réduire davantage l’incertitude. De toute évidence, nous devons faire des efforts dans les quantités d’entrée qui ont la plus grande part de l’incertitude afin que la mesure d’absorbance de l’échantillon et la décomposition contaminante possible est une chose à prendre en compte. Afin que tous les efforts soient consacrés à d’éventuelles interférences spectrales, d’éventuelles instabilités du colorant, d’éventuelles contaminations, d’éventuelles décompositions. Cependant dans l’ensemble, ce qui peut être dit de cette procédure, est qu’elle peut déjà être considérée comme assez optimisée. Et comment voyons-nous encore cela s’il y a plusieurs sources d’incertitude, qui sont d’une ampleur comparable, cela signifie que la procédure est assez optimisée. S’il n’y avait qu’une seule source d’incertitude avec, voyons, une contribution de 85% alors nous pourrions dire que oui cette quantité d’entrée particulière nécessite beaucoup d’attention et nous ne pouvons pas appeler notre procédure de mesure, optimisée. |
Conclusions 1
http://www.uttv.ee/naita?id=17647
|
We have now seen one uncertainty estimation of a chemical analysis procedure from the very beginning to the very end involving all the steps that there are in chemical analysis. And in broad terms, if we use modelling approach all the uncertainty estimations go via the same steps as given here, although oftentimes the steps can be either more voluminous or may be smaller than they were here. And in some cases, some of the steps may be omitted but the general workflow will always be the same. |
Nous avons désormais vu une estimation de l’incertitude d’une procédure d’analyse chimique du tout début à la fin impliquant toutes les étapes de l’analyse chimique. Et en termes généraux, si nous utilisons l’approche de modélisation toutes les estimations d’incertitudes passent par les mêmes étapes comme données ici, bien que souvent les étapes peuvent être plus volumineuses ou plus petites qu’elles ne l’étaient ici. Et dans certains cas, certaines étapes peuvent être omises mais le flux de travail général sera toujours le même. |
Conclusions 2
http://www.uttv.ee/naita?id=18096
|
What we just saw was a typical uncertainty estimation in chemical analysis in an instrumental analysis procedure. And the uncertainty sources like these that we had here, possible interferences, calibration graph, possibility composition, they are very often found in chemical analysis. On the other hand, as we saw several of the important uncertainty components, we just estimate based on experience or long-term results from the laboratory. And this also is fairly typical for chemical, analysis experience and an expert opinion oftentimes are indispensable for getting realistic uncertainty estimates in chemistry. And the worst that could be done would be just leaving out of consideration the uncertainty sources that cannot be found, some, by some fully rigorous approach or that can be found by some fully rigorous approach only with very large working effort. |
Ce que nous venons de voir était une estimation typique de l’incertitude dans l’analyse chimique dans une procédure d’analyse instrumentale. Et les sources d’incertitude comme celles que nous avions ici, les interférences possibles, le graphique de calibration, les possibilités de composition, elles sont très souvent retrouvées en chimie analytique. D’un autre côté, comme nous avons vu plusieurs des composantes importantes de l’incertitude, nous nous contentons d’estimations basées sur l’expérience ou les résultats à long terme du laboratoire. Et ceci est également assez typique pour les produits chimiques, une expérience d’analyse et des avis d’experts sont souvent indispensables pour obtenir des estimations réalistes de l’incertitude en chimie. Et le pire qui pourrait être fait serait de ne pas prendre en considération les sources d’incertitude qui ne peuvent pas être trouvées, certaines par des approches complètement rigoureuses ou qui ne peuvent être trouvées par des approches complètement rigoureuses seulement avec un effort de travail très important. |
Autoévaluation sur cette partie du cours : Test 9.9A ; Test 9.9B ; Test 9.9C
11. Approche par validation dite de laboratoire unique
Résumé : Cette section explique l'approche dite de validation par un laboratoire unique. Nous nous pencherons sur la formalisation de cette approche publiée par Nordtest. [1] C'est pourquoi, dans ce cours, cette approche est souvent appelée "l'approche Nordtest". L'approche de validation par un laboratoire unique, contrairement à l'approche de modélisation du GUM de l'ISO, n'approfondit pas la procédure de mesure et ne tente pas de quantifier individuellement toutes les sources d'incertitude. Au lieu de cela, les sources d'incertitude sont quantifiées en grands "lots" via des composantes qui prennent en compte un certain nombre de sources d'incertitude. La plupart des données utilisées proviennent de la validation de la procédure analytique. C'est la raison pour laquelle le mot "validation" figure dans le nom de l'approche. Ce type d'approche est aussi parfois appelé approche "descendante".
Les sections 10.1 à 10.3 présentent l'approche Nordtest étape par étape et expliquent la manière d'obtenir les données nécessaires. La section 10.4 donne une "feuille de route" de l'approche Nordtest. La section 10.5 présente un exemple pratique d'application de l'approche Nordtest dans le cas de la détermination de l'acrylamide dans les collations par chromatographie liquide et spectrométrie de masse (LC-MS).
- 10.1. Principes
- 10.2. Composantes d’incertitude associées aux effets aléatoires
- 10.3. Composantes d’incertitude associées aux effets systématiques
- 10.4. Feuille de route
- 10.5. Exemple pratique : détermination de la teneur en acrylamide dans des casse-croutes par LC-MS
***
[1] Handbook for Calculation of Measurement Uncertainty in Environmental Laboratories. B. Magnusson, T. Näykki, H. Hovind, M. Krysell. Nordtest technical report 537, ed. 3. Nordtest, 2011. Disponible en ligne sur http://www.nordtest.info/index.php/technical-reports/item/handbook-for-calculation-of-measurement-uncertainty-in-environmental-laboratories-nt-tr-537-edition-3.html
***
Les diapositives présentées dans cette section sont disponibles ici :
11.1. Principes
https://sisu.ut.ee/measurement/101-principle-absolute-and-relative-quantities
Dans l’approche Nordtest, l’incertitude est considérée comme due à deux composantes :
- La composante de reproductibilité intra-laboratoire (précision intermédiaire). Cette composante d’incertitude prend en compte toutes les sources d’incertitude aléatoires à long terme (c’est-à-dire plusieurs mois, de préférence un an). Ainsi, certaines sources d’incertitude qui sont systématiques en une journée deviendront aléatoires à long terme.
- La composante de biais. Cette composante prend en compte les effets systématiques qui provoquent un biais à long terme (mais pas ceux qui provoquent simplement un biais au cours d’une journée donnée). Le biais à long terme peut être considéré comme la somme du biais de la procédure (biais inhérent à la nature de la procédure) et du biais de laboratoire (biais causé par la façon dont la procédure est mise en œuvre en laboratoire).
Introduction à l'estimation de l'incertitude basée sur des données de validation et de contrôle qualité (l'approche Nordtest)
http://www.uttv.ee/naita?id=17909
| Let us now look at the uncertainty estimation approach which is based on validation and quality control data. And it is also quite often called the Nordtest approche and i've been using also this name throughout this course. So in this approach all the effects that contribute to the uncertainty are divided into random effects and systematic effects, and those random and systematic effects are then quantified separately. And in very broad terms and in a simplified way we could say that the main equation of this approach looks like this. The combined standard uncertainty of the result is found by combining the overall uncertainty arising from random effects and the overall uncertainty arising from the possible systematic effects. I will explain later on why this "possible" is put here and why it is in italic. It is now also very important to mention that both these random and systematic effects, they are taken or they are meant to be found at long-term level. Meaning the effects that are random are random during the time period of several months, and the same goes about the systematic effects. So the time frame that we are looking here is several months at least preferably year. | Regardons maintenant l'approche de l'estimation de l'incertitude qui est basée sur des données de validation et de contrôle de la qualité. Et cette approche est souvent appelée l'approche Nordtest et j'ai aussi utilisé ce nom tout au long de ce cours. Donc dans cette approche, tous les effets qui contribuent à l'incertitude sont partagés entre effets aléatoires et effets systématiques, et ces effets aléatoires et systématiques sont ensuite quantifiés séparément. Et dans des termes très généraux et dans une manière très simplifée nous pourrions dire que l'équation mathématique de cette approche ressemble à cela. L'incertitude-type composée de ce résultat est trouvée en combinant l'incertitude totale provenant des effets aléatoires et l'incertitude totale provenant des effets systématiques possibles. Je vais expliquer plus tard pourquoi ce "possible" est mis ici et pourquoi il est en italique. Il est maintenant également très important de mentionner que ces deux effets, aléatoires et systématiques, sont pris ou censés être trouvés sur le long terme. Cela signifie que les effets qui sont aléatoires sont aléatoires pendant une période de temps de plusieurs mois, et c'est la même chose pour les effets systématiques. Donc la période que nous regardons ici est au moins plusieurs mois, de préférence une année. |
L’équation principale de l’approche Nordtest est :
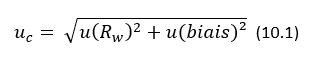
Ici u (Rw) représente la composante de reproductibilité intra-laboratoire de l'incertitude et u (biais) représente la composante d'incertitude en tenant compte des biais possibles. L'incertitude de mesure résultante uc n'est pas directement liée à un résultat spécifique, car elle est calculée à l'aide des données des mesures précédentes. On peut donc dire que l'incertitude obtenue avec l'approche Nordtest caractérise la procédure d'analyse plutôt qu'un résultat concret. Si l'incertitude d'un résultat concret est nécessaire, elle est affectée au résultat.
Pour cette raison, il est nécessaire de décider s'il faut exprimer l'incertitude en termes absolus (c'est-à-dire en unités de la quantité mesurée) ou en termes relatifs (c'est-à-dire en tant que rapport d'incertitude à la valeur de la quantité mesurée ou en pourcentage de la valeur mesurée). Les règles de bases :
- À de faibles concentrations (près de la limite de détection, niveau de trace), utiliser des incertitudes absolues
L'incertitude ne dépend pas beaucoup du niveau d'analyte
- À des concentrations moyennes et supérieures, utiliser des incertitudes relatives
L'incertitude est à peu près proportionnelle au niveau d'analyte
- En général : utilisez ce qui est le plus constant avec une concentration changeante
L'équation principale de l'approche Nordtest. Quantités absolues et relatives
http://www.uttv.ee/naita?id=17911
| The equation on the previous slide can be rewritten as you can see here on this line and this equation now comes directly from the Nordtest guide and also we are using throughout this explanation the terminology and the symbols that are used directly in the Nordtest guide. So the "uRw" component takes into account the long term random effects and it is actually the same thing as within lab long term reproducibility which we have seen already in a previous lecture. And secondly the "u(bias)" takes into account the possible systemic effects in long term and it is also interesting and important to note that this equation now can be used both with absolute and with relative values. And whether we use absolute values or whether we use relative value we have to determine based on the concrete situation. And this situation arises from the fact that the standard uncertainty that we find according to this approach is by itself not linked to any particular measurement result, in fact, we assign it to a result that we want to present to our customer. Whether we use absolute or relative quantities, depends on several things, but let me give you a rule of Thumb which one to use. And the rule of Thumb goes as follows, if your concentrations, if the contents of the analyte in the sample are very low, so you are working near the detection or near the quantification limit, then uncertainty is not very much dependent on the analyte level and then absolute quantities are more appropriate. If however you work at medium concentrations or at high concentrations then the uncertainty in chemical analysis very often is roughly proportional to the analyte level. This proportionality usually is not strict, so if you look at the uncertainties at different concentration levels then they differ slightly, but nevertheless the relative uncertainty stays more or less constant while the absolute uncertainty increases with the increasing concentration. And the general rule here would be whichever is more constant is better to use, meaning if we change in concentration, the absolute uncertainty remains constant then you should use absolute uncertainties. If we change in concentration, the relative uncertainty remains constant then you should use the relative uncertainty. | L'équation sur la diapositive précédente peut être réécrite comme vous pouvez le voir ici sur cette ligne. Cette équation provient directement du guide Nordtest et aussi nous utilisons tout au long de l'explication la terminologie et les symboles qui sont utilisés directement dans le guide Nordtest. Donc l'élément "u(Rw)" prend en compte les effets aléatoires de longue durée et c'est en fait la même chose que la reproductibilité intra-laboratoire à long terme que nous avons déjà vu dans la séance précédente. Et deuxièmement le "u(biais)" prend en compte les effets systématiques possibles à long terme et c'est aussi intéressant et important de noter que cette équation maintenant peut être utilisée aussi bien avec des valeurs absolues et relatives. Et que nous utilisons des valeurs absolues ou des valeurs relatives, nous devons le déterminer en fonction du contexte concret. Et cette situation résulte du fait que l'incertitude type que nous trouvons d'après cette approche est en elle-même non liée à un résultat de mesure particulier, en effet, nous l'attribuons à un résultat que nous voulons présenter à notre client. Le fait d'utiliser des quantités absolues ou relatives, dépend de plusieurs choses, mais laissez-moi vous donner une règle de Thumb que nous devons utiliser. La règle de Thumb est la suivante, si vos concentrations, si les teneurs en analyte dans l'échantillon sont très faibles, donc vous travaillez proche de la limite de détection ou de quantification, alors l'incertitude ne dépend pas beaucoup de la teneur en analyte et donc les quantités absolues sont plus appropriées. Si par contre vous travaillez à des concentrations moyennes ou à des hautes concentrations alors l'incertitude dans les analyses chimiques est très souvent à peu près proportionnelle au niveau d'analyte. Cette proportionnalité n'est habituellement pas stricte, donc si vous regardez les incertitudes aux différents niveaux de concentration alors ils diffèrent légèrement, mais néanmoins l'incertitude relative reste plus ou moins constante pendant que l'incertitude absolue croît avec l'augmentation de la concentration. Et la règle générale ici serait qu'il est préférable d'utiliser celle qui est la plus constante. Ce qui signifie que si nous changeons en concentration et l'incertitude absolue reste constante alors vous devriez utiliser les incertitudes absolues. Si nous changeons en concentration et l'incertitude relative reste constante alors vous devriez utiliser l'incertitude relative. |
Aperçu de la mise en œuvre pratique de l'approche Nordtest
http://www.uttv.ee/naita?id=17912
| Let us look now how to implement this approach in practice and this slide here presents the steps of uncertainty estimation using the Nordtest approach. Some of the steps are the same as they were in the modeling approach but some are different so as it was with the modeling approach we start by specifying the measurement but then here we don't have the model part it doesn't mean that we don't need any model we do need the model because we need to calculate the results somehow when this goes via the measurement model however the model is not directly a part of the uncertainty estimation approach as such so in the second and third step we quantify separately the "urw" component or the so-called, component taking into account the random effects and the component taking into account the possible bias and then both components need to be converted to the standard uncertainties and the remaining quadratic curves exactly as it was in the modeling approach. We can calculate from thesecomponents the combined standard uncertainty and finally we calculate the expanded answer. | Regardons maintenant comment mettre en oeuvre cette approche dans la pratique et cette diapositive ici présente les étapes de l'estimation de l'incertitude en utilisant l'approche Nordtest. Certaines de ces étapes sont les mêmes que celles de l'approche par modélisation mais certaines sont différentes de celles de l'approche par modélisation. Nous commençons par spécifier la mesure mais après ici nous n'avons pas la partie modélisée, cela ne veut pas dire que nous n'avons pas besoin d'un modèle, nous en avons besoin car nous devons calculer les résultats d'une certaine façon quand cela va via le modèle de mesure. Par contre le modèle ne fait pas directement partie de l'approche de l'estimation de l'incertitude en tant que tel. Donc dans les deuxième et troisième étapes nous quantifions séparément l'élément "u(Rw)" ou aussi appelé l'élément qui prend en compte les effets aléatoires, et l'élément prenant en compte les biais possibles. Et ensuite les deux éléments doivent être convertis en incertitudes-types et des courbes quadratiques restantes exactement comme c'était dans l'approche par modélisation. Nous pouvons calculer à partir de ces éléments l'incertitude-type composée et finalement nous calculons la réponse étendue. |
Les principales étapes du processus d'évaluation de l'incertitude de mesure avec l'approche Nordtest :
- Spécifier le mesurande
- Quantifier la composante Rw u(Rw)
- Quantifier la composante de biais u(biais)
- Convertir les composants en incertitudes-types u(x)
- Calcul de l'incertitude-type composée uc
- Calcul de l'incertitude élargie U
***
[1] Un exemple typique est le titrage si un nouveau titrant est préparé chaque semaine. Dans une semaine donnée, la concentration de titrant est un effet systématique, mais à long terme, elle devient aléatoire, car de nombreux lots de titrant seront impliqués. Un exemple similaire typique est le graphique d'étalonnage s'il est préparé quotidiennement : chaque jour, le biais possible d'étalonnage est un effet systématique, mais à long terme, il devient aléatoire.
11.2. Composantes d'incertitude associées aux effets aléatoires
https://sisu.ut.ee/measurement/102-estimating-uncertainty-component-accounting-random-effects-using-data-are-available
Estimation de la composante de reproductibilité intra-laboratoire de l'incertitude
http://www.uttv.ee/naita?id=17913
| And we can now look at more detailed how exactly we find the values of these uncertainty components. So the u(Rw) takes into account long-term variation of results within laboratory. And it is measured in such a way that you have the same sample. The sample should be as similar as possible to the test samples by its matrix, by concentration, by homogeneity. And you should have enough of that sample so that you can carry out measurements during long time period. And the sample of course has to be stable enough so that it doesn't decompose and that the analyte concentration doesn't change with time. Then obviously the same lab and the same procedure. But now as over the long time period, different things can happen. Of course the measurements are carried out on different days. If, in your laboratory, different persons do that same measurement, then on different days obviously it can be different people who do the measurement. There are different reagent batches, there can be different calibration graphs, different detectors if the detector of the instrument is changed sometimes, different LC or GC columns if it is a chromatographic method and columnist chains etc. So this list can be made much longer and whatever needs to be changed during the normal routine work of your laboratory has to be changed. And extremely importantly, this measurement has to include all steps including sample preparation so that sample really has to be a real sample, not the solution made of the real sample, so that on each and every of those different days over long time period also sample preparation is carried out separate. And let us just for a moment remind again this scheme here which I presented already in an earlier lecture. This, within lab long term reproducibility (SRw), is always larger than repeatability (Sr) because it takes in several effects that within a single day would be systematic effects, but in long term, become random effects. On the other hand, SRw still is smaller than the combined standard uncertainty (Uc) because the combined standard uncertainty also has to include systematic effects that remain in systematic even over this long time period. So let us summarize : u(Rw) is found as long term within laboratory standard deviation, there has to be a control sample, the analysis has to cover the whole analytical process including sample preparation. And if this particular procedure is used with different matrixes or at very different concentration levels, then for different matrixes and different concentration levels, ideally you should have different control samples. | Nous pouvons maintenant examiner plus en détail comment trouver exactement les valeurs de ces composantes d'incertitude. u(Rw) tient compte de la variation à long terme des résultats au sein du laboratoire. Il est mesuré de telle sorte que l'on ait le même échantillon. L'échantillon doit être aussi semblable que possible aux échantillons d'essai par sa matrice, sa concentration, son homogénéité. Vous devez avoir suffisamment d'échantillon pour pouvoir effectuer des mesures sur une longue période. L'échantillon doit bien sûr être suffisamment stable pour ne pas se décomposer et que la concentration de l'analyte ne change pas avec le temps. Il faut aussi le même laboratoire et la même procédure. En revanche, sur une longue période, différentes choses peuvent se produire. Bien sûr les mesures sont effectuées sur différents jours. Si dans votre laboratoire, différentes personnes font cette même mesure, alors forcément à des jours différents, ce sont des personnes différentes qui effectuent la mesure. Il y a différents lots de réactifs, il peut y avoir différents graphiques de calibration, différents détecteurs (si le détecteur de l'instrument est parfois changé), différentes colonnes LC ou GC s'il s'agit d'une méthode chromatographique et de chaînes de colonnes etc. Cette liste peut être bien plus longue et tout ce qui a besoin d'être modifié durant le travail de routine normal de votre laboratoire doit être changé. Extrêmement important : cette mesure doit comprendre toutes les étapes, y compris la préparation d'échantillon afin qu'il soit vraiment un échantillon réel, pas une solution faite de l'échantillon réel, de sorte que pour chacun de ces jours différents sur une longue période, la préparation de l'échantillon soit aussi effectuée séparément. Revenons encore un instant sur ce schéma ici que j'ai déjà présenté dans un cours précédent. Ici, la reproductibilité à long terme en laboratoire (SRw), est toujours plus grande que la répétabilité (Sr) car elle prend en compte plusieurs effets qui, en un seul jour seraient des effets systématiques mais à long terme, deviennent des effets aléatoires. D'un autre côté, SRw est toujours plus petit que l'incertitude-type composée (Uc) parce que l'incertitude-type composée doit aussi inclure les effets systématiques, qui restent systématiques même sur cette longue période de temps. Résumons : u(Rw) est trouvé comme l'écart-type de laboratoire à long terme. Il doit y avoir un échantillon de contrôle, l'analyse doit couvrir l'ensemble du processus analytique dont la préparation d'échantillon. Si cette procédure particulière est utilisée avec différentes matrices ou à des niveaux de concentration très différents, alors pour différentes matrices et différents niveaux de concentration, vous devez idéalement avoir différents échantillons de contrôle. |
La reproductibilité intra-laboratoire (fidélité intermédiaire, SRW notée u(Rw) dans le guide Nordtest) prend en compte toutes les sources d’incertitude, qui sont aléatoires à long terme (par exemple plusieurs mois, de préférence un an). Donc, certaines sources d’incertitude qui sont systématiques en une journée deviendront aléatoires à long terme.
Il s’agit de l’un des points clés de l’approche de validation intra-laboratoire de l’estimation de l’incertitude : essayer de tenir compte du plus grand nombre possible de sources d’incertitude par le biais d’effets aléatoires. Il s’agit également d’un avantage important de l’approche, car, en règle générale, l’estimation des effets aléatoires peut être effectuée de façon plus fiable que l’estimation des effets systématiques. La raison en est que pour évaluer les effets aléatoires, il n’est pas nécessaire d’avoir une valeur de référence, mais pour évaluer les effets systématiques il le faut (voir la section 10.3).
La façon la plus simple de trouver u(Rw) est de prendre un certain nombre de mesures répétées d’un échantillon de contrôle, organisées, par exemple, sous forme d’un graphique de contrôle. Il est également possible d’utiliser l’approche de l’écart-type groupé comme expliqué à la section 6. Si cela est fait, l’u(Rw) peut être trouvé sur la base de plusieurs échantillons de contrôle différents, de sorte qu’il soit une valeur moyenne de tous.
Le nombre de valeurs utilisées pour l’évaluation de u(Rw)doit être suffisamment important. Une estimation initiale de u(Rw) peut être obtenue avec 10-15 valeurs, mais par la suite plus de données devraient être collectées. Plus important encore, la période pendant laquelle les données sont collectées, doit être suffisamment longue (au moins plusieurs mois, de préférence environ un an) pour que toutes les sources de variabilité de la procédure soient prises en compte. Ainsi, 10 valeurs recueillies sur une période de cinq mois est une meilleure option que 20 valeurs recueillies pendant 1,5 mois. Si les mesures sont effectuées avec le même échantillon de contrôle, alors il doit être disponible en quantité suffisante et doit être stable pendant la période. Le tableau 6.2 de la section 6 donne un aperçu compact des exigences de la détermination de u(Rw) (sRW).
Selon la situation, u(Rw) peut être utilisé comme valeur absolue ou relative.
Il est important de souligner que u(Rw) doit être estimé séparément pour différentes matrices et différents niveaux de concentration.
Autoévaluation sur cette partie du cours : Test 10.2
11.3. Composantes d'incertitude associées aux effets systématiques
Estimation des composantes d'incertitude dues à des biais possibles
http://www.uttv.ee/naita?id=17910
| Let us now look at the bias component. And, let me explain now why I've put in several places, here, the word "possible". The point is that determining the true bias of the procedure is not easy. It needs a lot of parallel measurements under carefully controlled conditions. And, if the number of measurements is not very large, then the bias estimate always will be mixed with random effects, so that the bias will also contain contributions from random effect. And, it can easily happen that in fact, the procedure does not have any bias at all. But, simply, we are not able to separate the systematic and random effects because we do not do enough measurements. And, making many many many parallel measurements can be impossible for practical reasons. Therefore, what often happens is that we don't really know if our procedure has a bias or not. But, we quantify our "non-knowledge" of the existence of bias by the u(bias) uncertainty component. Now, if we were looking at the random component, the RW component, then I did not specify anything about the sample. The sample there could be just any sample. Here, it's different. For finding bias, we certainly need a reference value. We've seen in an earlier lecture that this bias actually is a numerical estimate or numerical quantification possibility of trueness. And, whenever we speak about trueness, we always need a reference value meaning. For bias determination, we now need samples or need materials where we know, at least from some kind of information, how much of the analyte is inside. And, in broad terms, there are four different possibilities how bias can be found. First of them, is that you have some sample and the sample is analyzed with your procedure, and then, within your reference procedure. And, that reference procedure preferably should have low uncertainty at high reliability. This is an excellent way but unfortunately, very often, such reference procedure is not available. And, therefore, it's unfortunately not very often used. The second one, which is actually also quite good, is that you buy certified reference materials. And, certified reference material means material which is very much like your real sample, but where the contents of certain analytes are known very reliably and they have been determined by expert laboratories using high level analytical techniques. And, now, the reference values of the certified reference materials are well suitable as reference values for bias determination. The next possibility is to participate in the interlaboratory comparison measurements. And, in the interlaboratory comparison measurements, the organizer always eventually announces the so-called consensus or reference value, so that, again, you will have a reference value for the sample. But, unfortunately, in most cases in interlaboratory comparisons, and in almost all cases where they have the so-called proficiency testing time, the reference values are found from the results of the participants. And, since there are always quite different levels of participants, and some of them are perhaps not so good, then these reference values or usually as they're called "consensus values of interlaboratory comparison measurements" cannot be called very reliable. And, if the interlaboratory comparisons are used for bias estimation, very often overestimated bias is the result. And, finally, it is also possible to estimate bias from spiking experiments, meaning you have some kind of sample in your laboratory, or even better, you have a sample matrix, blank matrix, where there is no analyte inside. And, then, you add a well-known quantity of the analyte into that matrix, and then, you determine by your analytical technique whether you get back as much as you added. And, the difference between how much you got and how much you added is the bias. Now, let me draw your attention to this word "repeated". Repetition is very important in bias determination, simply because, otherwise, your bias estimate will contain a fair share of random effects in it. Therefore, always for determining bias, you have to carry out several repeated determinations. And, furthermore, bias can change from matrix to matrix, and also bias can be dependent on concentration. Therefore, ideally you should use several different reference materials, several different proficiency test results, to estimate your bias so that you finally would get some kind of averaged bias of your procedure, and that you could avoid that your bias will be grossly underestimated, or grossly overestimated. Let us see now, how we calculate this bias component, the main equation is presented here. So your bias is found as a combination of to uncertainty comports. This RMS bias accounts for the average bias in your laboratory. I will show on the coming slides how it is calculated. It basically is the average bias value and this component u( C ref) takes into account the average uncertainty of the reference values. So as you remember from earlier lectures, our reference values are different from true values in that they have uncertainties and these uncertainties are counted for by this component. And now, here are even some of the equations that you are going to need. So each and every separate bias value is calculated like this. So it's the concentration found in your lab, minus the reference concentration, will give a single bias value whereby this Clabi should be determined from repeated measurements. So that each of these bias values, in turn, should come from repeated measurements. And then you should get several bias values, preferably from several different reference materials or several different internal comparisons. Finally the RMS bias is found as follows : You take all your bias estimates that you obtained, You take all of them to the square. You sum all of them up, divide by the number how many bias estimates you had. And finally take square root. And averaging via this procedures you see here, is called averaging a root-mean- square approach and this is where the RMS comes from. Similar situation holds with this u(Cref). If the bias determination is carried out with certified reference materials, then this u(Cref) can be found from a certificate of certified reference material. If bias determination is carried out by inter laboratory comparisons and if the values the consensus values on the inter laboratory comparisons are determined from the participant results, which is the most common way, then the u(Cref ) can be estimated as follows. This here is now the standard deviation of the participant results, in the interlaboratory comparison and this here is the number of participants in this particular interlaboratory comparison. And for every interlaboratory comparison, this u(Cref i) is found and separately u(Cref i) also found for different reference materials or for other approaches that I mentioned in the previous line. And finally u(Cref i) values are averaged it averaged exactly the same way as we saw here by the root-mean-square procedure, meaning all of them are separately taken to the square. Squares are summed and divided by the number of u(Cref) estimates that you have. And this is the average uncertainty of the reference value. And let me once more stress, that if the number of bias estimates, is too small, if this end is too small, then our bias estimate will include a large share of random effects in it. And the bias can be overestimated. if it happens and this is an unfortunate situation, that we have only one single certified reference material for bias estimation. But we can do several bias determinations with it. Then the u(bias) formula changes a little bit. So this RMS bias squared and u(Cref) remain the same, but in addition we have to bring in additional term, which is the standard deviation of the bias values obtained and this is how many bias determinations were carried out. So such additional term has to be brought in, if only one single certified reference material is. And again as it was in the case of the random component, the urw, preferably the bias should be estimated separately for different matrixes. And also if very different concentrations are used, then also separately for different concentration levels. | Regardons maintenant la composante du biais. Et, laissez-moi vous expliquer pourquoi j'ai mis à plusieurs endroits, ici, le mot "possible". Le fait est que déterminer le véritable biais de la procédure n'est pas facile. Cela nécessite de nombreuses mesures parallèles dans des conditions soigneusement contrôlées. Et, si le nombre de mesures n'est pas très grand, alors l'estimation du biais sera toujours mélangée avec des effets aléatoires, de sorte que le biais contiendra également les contributions des effets aléatoires. Et, il peut facilement arriver qu'en fait, la procédure ne possède aucun biais du tout. Mais, tout simplement, nous ne sommes pas capables de séparer les effets systématiques et aléatoires parce que nous ne faisons pas suffisamment de mesures. Et, faire de nombreuses et nombreuses mesures parallèles peut être impossible pour des raisons pratiques. Par conséquent, ce qui arrive souvent, est que nous ne savons pas vraiment si notre procédure a un biais ou non. Mais, nous quantifions notre "non-connaissance" de l'existence d'un biais par la composante d'incertitude u(biais). Maintenant, si nous examinons la composante aléatoire, la composante RW, je n'ai rien spécifié à propos de l'échantillon. L'échantillon pourrait être n'importe quel échantillon. Ici, c'est différent. Pour trouver le biais, nous avons certainement besoin d'une valeur de référence. Nous avons vu dans une conférence précédente que ce biais est en fait une estimation numérique ou une possibilité de quantification numérique de la justesse. Et, chaque fois que nous parlons de justesse, nous avons toujours besoin de la signification de la valeur de référence. Pour la détermination du biais, nous avons besoin d'échantillons ou de matériaux dont nous connaissons, au moins à partir d'une certaine information, combien d'analyte se trouve à l'intérieur. Et, en termes généraux, il existe quatre différentes possibilités de trouver un biais. La première, c'est que vous disposez d'un échantillon qui est analysé selon votre procédure, et puis, selon votre procédure de référence. Et, cette procédure de référence devrait, de préférence, avoir une faible incertitude avec une haute fiabilité. Ceci est un excellent moyen mais malheureusement, très souvent, une telle procédure de référence n'est pas disponible. Et, par conséquent, elle n'est malheureusement pas très souvent utilisée. La deuxième, qui est en fait également assez correcte, est que vous achetez des matériaux de référence certifiés. Et, un matériau de référence certifié signifie un matériau qui ressemble fortement à votre échantillon réel, mais dont le contenu de certains analytes est connu de manière fiable, et a été déterminé par des laboratoires experts utilisant des techniques analytiques de haut niveau. Et, maintenant, les valeurs de référence des matériaux de référence certifiés sont bien adaptées comme valeurs de référence pour la détermination du biais. La prochaine possibilité est de participer aux mesures de comparaison inter-laboratoires. Et, dans les mesures de comparaison inter-laboratoires, l'organisateur annonce toujours en fin de compte la valeur dite de consensus ou de référence, de sorte que, là encore, vous aurez une valeur de référence pour l'échantillon. Mais, malheureusement, dans la plupart des cas de comparaisons inter-laboratoires, et dans presque tous les cas où ils ont ce que l'on appelle le temps d'essai d'aptitude, les valeurs de référence sont trouvées à partir des résultats des participants. Et, puisqu'il y a toujours des niveaux de participants très différents, et, que certains ne sont peut-être pas si bons, alors ces valeurs de référence ou généralement appelées "valeurs de consensus des mesures de comparaison d'inter-laboratoires" ne peuvent pas être qualifiées de très fiables. Et, si les comparaisons d'inter-laboratoires sont utilisées pour l'estimation du biais, très souvent le biais surestimé est le résultat. Et, enfin, il est également possible d'estimer le biais issu des expériences de dopage c'est-à-dire que vous avez un certain type d'échantillon dans votre laboratoire, ou mieux encore, vous avez une matrice d'échantillon, une matrice vierge, dans laquelle il n'y a pas d'analyte. Et, ensuite, vous ajoutez une quantité bien connue de l'analyte dans cette matrice, et puis, vous déterminez par votre technique analytique si vous récupérez autant que ce que vous avez ajouté. Et, la différence entre ce que vous avez obtenu et ce que vous avez ajouté est le biais. Maintenant, permettez-moi d'attirer votre attention sur ce mot "répété". La répétition est très importante dans la détermination du biais, simplement parce que, sinon, votre estimation du biais contiendra une part importante d'effets aléatoires. Par conséquent, pour toujours déterminer le biais, vous devez effectuer plusieurs déterminations répétées. Et, de plus, le biais peut changer d'une matrice à l'autre, et le biais peut également dépendre de la concentration. Donc, vous devriez idéalement utiliser plusieurs matériaux de référence différents, plusieurs résultats de tests de compétences différents, pour estimer votre biais afin que vous obteniez finalement une sorte de biais moyen de votre procédure, et que vous puissiez éviter que votre biais soit largement sous-estimé, ou largement surestimé. Voyons maintenant, comment nous calculons cette composante du biais, l’équation principale est présentée ici. Votre biais se présente comme une combinaison de facteurs d'incertitude. Ce biais RMS représente le biais moyen dans votre laboratoire. Je vais montrer sur les prochaines diapositives comment il est calculé. Il s'agit essentiellement de la valeur de biais moyenne et cette composante u(Cref) tient compte de l'incertitude moyenne des valeurs de référence. Ainsi, comme vous vous en souvenez, lors de conférences précédentes, nos valeurs de référence sont différentes des vraies valeurs, dans le sens qu'elles ont des incertitudes et ces incertitudes sont comprises par cette composante. Et maintenant, voici, ici, quelques-unes des équations dont vous aurez besoin. Ainsi, chaque valeur de biais distincte est calculée comme ceci. Donc, c'est la concentration trouvée dans votre laboratoire, moins la concentration de référence, qui donnera une valeur de biais unique par lequel ce C(labi) devrait être déterminé à partir de mesures répétées. Ainsi, chacune de ces valeurs de biais, à leur tour, devront provenir de mesures répétées. Puis vous devriez obtenir plusieurs valeurs de biais, de préférence à partir de plusieurs matériaux de référence différentes ou plusieurs comparaisons internes différentes. Enfin, le biais RMS est trouvé comme suit : vous prenez toutes les estimations de biais que vous avez obtenues, vous les mettez toutes au carré. Vous les additionnez toutes, et divisez par le nombre d'estimations de biais que vous aviez. Et pour finir mettez le tout sous la racine carré. Et le calcul de la moyenne par ces procédures que vous voyez ici, s'appelle la méthode de la moyenne quadratique et c'est de là que vient le RMS. Une situation similaire se présente avec cette u(Cref). Si la détermination du biais est effectuée avec des matériaux de référence certifiés, alors cette u(Cef) peut être trouvée à partir d'un certificat de matériel de référence certifié. SI la détermination du biais est effectuée par des comparaisons inter-laboratoires et si les valeurs de consensus sur les comparaisons inter-laboratoires sont déterminées à partir des résultats des participants, ce qui est la méthode la plus courante, alors le u(Cref) peut être estimé comme suit. Il s'agit ici de l'écart type des résultats des participants, dans la comparaison entre laboratoires et il s'agit ici du nombre de participants à cette comparaison inter-laboratoire particulière. Et pour chaque comparaison inter-laboratoire, ce u(Crefi) est trouvé et séparément u(Crefi) est également trouvé pour différents matériaux de référence ou pour d'autres approches que j'ai mentionnées précédemment. Enfin les valeurs u(Crefi) sont moyennées exactement de la même façon que nous l'avons vu ici par la procédure de la moyenne quadratique, ce qui signifie que toutes sont prises séparément et portées au carré. Les carrés sont additionnés et divisés par le nombre d'estimations d'u(Cref) que vous avez. Et c'est l'incertitude moyenne de la valeur de référence. Permettez moi de souligner encore une fois, qui si le nombre d'estimations de biais est trop faible, si cette extrémité est trop petite, alors notre estimation du biais inclura une grande part d'effets aléatoires. Et le biais peut être surestimé. Si cela se produit et c'est une situation regrettable, nous n'avons alors qu'un seul matériel de référence certifié pour l'estimation du biais. Mais nous pouvons faire plusieurs déterminations avec lui. Ensuite, la formule de u(biais) change un peu. Donc ce RMS biais au carré et u(Cref) restent les mêmes, mais en plus nous devons apporter un terme supplémentaire, qui est l'écart type des valeurs de biais obtenues et c'est le nombre de déterminations de biais qui ont été effectuées. Il faut donc prévoir un terme supplémentaire, si un seul matériel de référence certifié est utilisé. Encore une fois, comme c'était le cas de la composante aléatoire, l' urw, le biais devrait être estimé de préférence séparément pour différentes matrices. Et aussi si des concentrations très différentes sont utilisées , puis également séparément pour différents niveaux de concentrations. |
La composante u(biais) prend en compte un éventuel biais de la procédure de mesure.
Il n’est pas facile de déterminer de manière fiable le biais de la procédure pour les raisons suivantes :
1. Si les effets aléatoires sont forts, cela nécessite un très grand nombre de mesures. Lorsqu'un nombre limité de mesures est effectué, l'estimation du biais contiendra toujours une contribution d'effets aléatoires, ce qui rendra l'estimation du biais artificiellement plus élevée. Encore plus - la procédure ne peut en fait avoir aucun biais.
2. Même avec des matrices formellement similaires, le biais peut différer en ordre de grandeur et même en signe (par exemple, lors de la détermination de pesticides dans différentes variétés de pommes, de la détermination des résidus de médicament dans le plasma sanguin de différents patients, etc.). Cela signifie qu'ayant déterminé le biais dans une variété de pommes, ce biais n'est pas automatiquement applicable pour une autre variété et nous pouvons parler d'incertitude dans l'application du biais à une autre variété.
3. Le biais est toujours déterminé par rapport à une valeur de référence, qui présente également une incertitude.
C'est pourquoi nous parlons d'un biais possible et la composante d'incertitude u (biais) quantifie notre connaissance limitée du biais.
Le biais fait référence à la différence entre notre valeur mesurée et une valeur de référence. Par conséquent, pour trouver u (biais), nous avons besoin d'un échantillon ou d'un matériau avec une valeur de référence. En termes généraux, il existe quatre possibilités différentes pour déterminer le u (biais) :
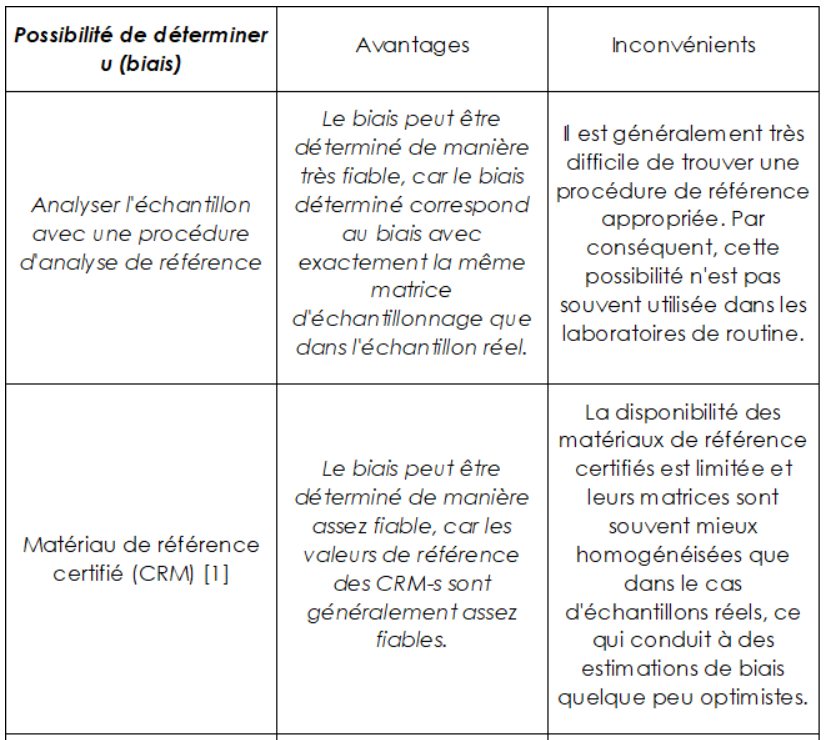
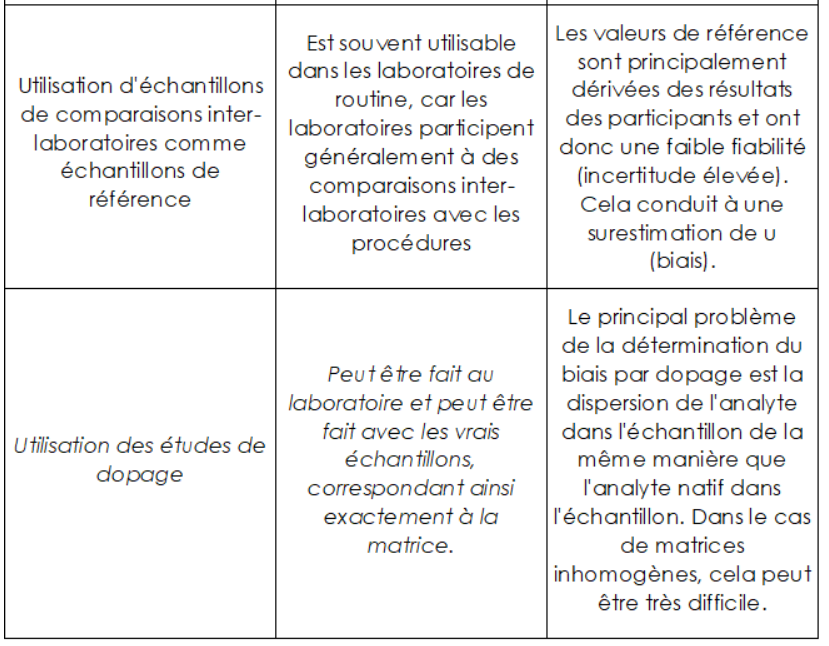
Dans le cas des quatre possibilités, il est essentiel d'inclure également la préparation des échantillons dans la détermination du biais.
Dans le cas des quatre possibilités, il est nécessaire de faire un nombre aussi grand que possible de mesures répétées afin de séparer le biais des effets aléatoires aussi efficacement que possible. Le biais change généralement d'une matrice à l'autre et est généralement différent à différents niveaux de concentration. Il est donc important d'utiliser plusieurs CRM, plusieurs comparaisons inter-laboratoires, etc.
Plus la fiabilité de la valeur de référence est faible, plus l'estimation u (biais) est élevée.
La composante u (biais) est trouvée selon l'équation suivante :
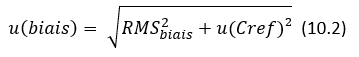
RMSbiais est le biais moyen (moyenne quadratique) et se trouve comme suit :
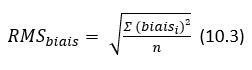
Où n est le nombre de déterminations de biais effectuées et chaque biais est le résultat d'une détermination de biais individuel et se trouve comme suit :
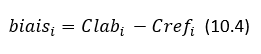
Où Clabi est une moyenne des résultats de la détermination de l'analyte dans l'échantillon de référence (par exemple dans le CRM) obtenu par le laboratoire et Crefi est la valeur de référence de l'échantillon de référence. Il est important que Clabi corresponde à un certain nombre de répliques.
u (Cref) est l'incertitude type moyenne des valeurs de référence des échantillons de référence et se trouve comme suit :
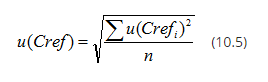
Ici u (Crefi) est l'incertitude-type de la xième valeur de référence. Dans le cas d'une analyse CRM, d'un dopage ou d'une analyse avec une procédure de référence, le u (Crefi) peut généralement être raisonnablement trouvé. Cependant, dans le cas de comparaisons inter-laboratoires où la valeur consensuelle des participants est utilisée comme valeur de référence, aucune incertitude fiable de la valeur de référence ne peut être trouvée. La meilleure estimation dans ce cas serait l'écart type de la valeur moyenne après élimination des valeurs aberrantes :
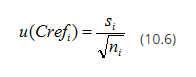
Ici, si est l'écart-type des participants à la xième inter-comparaison après élimination des valeurs aberrantes et ni est le nombre de participants (là encore après élimination des valeurs aberrantes) à la xième inter-comparaison.
Dans le cas particulier, si un certain nombre de déterminations de biais ont été effectuées à l'aide d'un seul CRM, l'équation 10.2 prend la forme suivante :
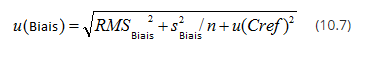
Où sbiais est l'écart-type des estimations de biais obtenues et n est le nombre d'estimations de biais obtenues.
Selon la situation, le u (biais) peut être utilisé comme valeur absolue ou comme valeur relative.
Enfin, il est important de souligner que la composante d'incertitude du biais doit être estimée séparément pour différentes matrices et différents niveaux de concentration.
***
[1] En termes simplifiés, le matériau de référence certifié est un matériau dans lequel le contenu de l'analyte (ou des analytes) est connu de manière fiable (le matériau a un certificat). Si la matrice du matériau de référence certifié est similaire à des échantillons réels (c'est-à-dire qu'il ne s'agit pas d'un composé pur ou d'une solution mais par exemple du lait en poudre, de la terre ou du plasma sanguin), nous l'appelons matériau de référence matriciel certifié.
Autoévaluation sur cette partie du cours : Test 10.3
11.4. Feuille de route
https://sisu.ut.ee/measurement/104-practical-example
Feuille de route d'estimation d'incertitudes en utilisant l'approche Nordtest
http://www.uttv.ee/naita?id=17914
| Let us look now at the roadmap of measurement uncertainty estimation using the Northtest approach. So I have collected all the formulas and calculations into this slide and we can now briefly go through them to get the big overview. Because otherwise, it can be difficult to follow all these calculations. So, we start by finding un biais so each individual bias value is found by numerous laboratory measurement results from which the reference value has been subtracted. And then,these are averaged into the RMSbias. Secondly, from either from the certificate, or from interlaboratory comparison data, we find the uncertainties of those reference values. And these are term averaged into the u(Cref) using again this root mean square approach. Now RMSbias and u(Cref) are combined into the over all possible bias estimates. And then, separately, we look at uncertainty due to random effects where we carry out determination of long term within laboratory standard deviation by using a control sample, or several control samples if necessary, that we analyze over a long time period. Several months or preferably one year. And when we found this u(Rw) and when we have u(bias), then these two we can eventually combine into the combined standard uncertainty of the result. Not too difficult. | Regardons maintenant la feuille de route de l'estimation de l'incertitude de mesures en utilisant l'approche Nordtest. J'ai donc rassemblé toutes les formules et les calculs dans cette diapositive et nous pouvons maintenant les parcourir brièvement pour obtenir une vue d'ensemble. Parce qu'autrement, il peut être difficile de suivre tous ces calculs. Donc, nous commençons par trouver un biais de sorte que chaque valeur de biais individuelle est trouvée par de nombreux résultats de mesure en laboratoire dont la valeur de référence a été soustraite. Et puis, ceux-ci sont moyennés dans les RMSbiais. Deuxièmement, soit à partir du certificat, soit à partir de données de comparaison inter-laboratoires, nous trouvons les incertitudes de ces valeurs de référence. Et ces termes sont moyennés dans le u(Cref) en utilisant à nouveau cette approche quadratique moyenne. Maintenant, RMSbiais et u(Cref) sont combinés dans toutes les estimations de biais possibles. Et puis, séparément, nous examinons l'incertitude due aux effets aléatoires où nous effectuons la détermination à long terme de l'écart-type en laboratoire en utilisant un échantillon de contrôle, ou plusieurs échantillons de contrôle si nécessaire, que nous analysons sur une longue période. Plusieurs mois ou de préférence un an. Et quand nous avons trouvé ce u(Rw) et lorsque nous avons u(biais), nous pouvons finalement combiner ces deux dans l'incertitude-type composée du résultat. Pas trop difficile. |
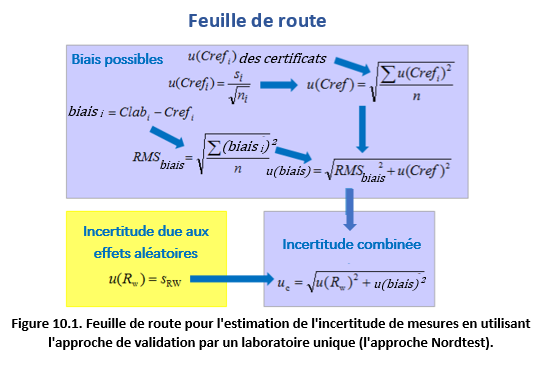
11.5. Exemple pratique : détermination de la teneur en acrylamide dans des casse-croutes par LC-MS
https://sisu.ut.ee/measurement/105-practical-example-uncertainty-estimation-using-nordtest-approach
L'approche Nordtest en pratique : Détermination de l'acrylamide dans des en-cas par LC-MS
http://www.uttv.ee/naita?id=18163
| Let us look now how the single lab validation approach for the Nordtest approach works in practice. And we will look at that on the example of determination of acrylamide in snacks by liquid chromatography mass spectrometry. Acrylamide is a toxic contaminant sometimes found in foods which are produced by heated processing: for example potato snacks, crisp bread, etc… And since it's a toxic compound and occasionally it's content in the snacks can reach quite high levels, its determination in such foods is very important. As I've said previously in the single lab validation approach, the uncertainty is calculated rather for the procedure for the analytical method than for the particular result. And what we will do now, we also will calculate it for the procedure and afterwards, apply it to this result. So the laboratory has found concentration level 998 micrograms per kilogram and this is our result for which we will eventually assign the uncertainty. Now for finding the bias and in fact also the reproducibility component of uncertainty, laboratory has two certified reference materials. One of them is potato chips and the other one is crisp bread. And for both of these there are reference values available and in the case of crisp bread this also has been used as a control sample by the laboratory. Meaning the laboratory has a sufficient amount of it and it has been analyzed many times over a long time period. And let us see now what are the data of these crms (Certified Reference Materials) and we can see the data here. These are the reference values of acrylamide. And these are the expanded uncertainties at k2 level. Let us look now at the data that the laboratory has for these certified reference materials. With crisp bread, a number of determination has been carried out on different days and all these values together lead to the laboratory mean value which can be compared with the certified reference value, and therefore used for bias determination. On the other hand since determination has been carried out during a long time, more than one year, these values also can be used for determining within lab long term reproducibility or the component accounting for the random effects. So that this certified reference materials can be used for both of these purposes. The other certified reference material has also been analyzed several times, leading to again this mean value. Meaning, we can use this value for bias determination. But for within lab long term reproducibility, we cannot use these results because all the determinations were done during a relatively short time period and only on two different days. Our uncertainty evaluation will proceed according to the roadmap that we have seen already earlier, which is given here.So from one of the CRM analysis we can get this uncertainty due to the random effects: within lab long term reproducibility, and secondly from both of them we can get the bias value. So both of them give us one single bias value which, there after, we average, both of them, also have, u(C ref) values meaning uncertainties on the reference values which we also average and then calculate u(bias). And finally u(Rw) and u(bias) are combined together into combine standard uncertainty estimate. How exactly these calculations are carried out we will look at the real calculation example with real data using Microsoft Excel. I have prepared here a calculation sheet where I have introduced already several things that we will need to calculate. And also I have introduced here the measurement result value. And secondly we have here the sheet where all the data are given: both the reference values and the uncertainties of both of the reference materials and also all the parallel determinations and the values that the laboratory has obtained. And we will now look at how these data that we have here on this sheet will be used for calculating the measurement uncertainty in the sheet. And we will start with the reproducibility component. As I said, we can use only the first certified reference material for reproducibility evaluation. And the reproducibility will be evaluated simply as the standard deviation of those within lab long-term measurements. So that the u(Rw) is actually equal to the standard deviation of the laboratory measurement. And of course we have to put here the units. So it's µg/kg. Now in this case, we will be using relative quantities for uncertainties calculation as we, as we saw, in one of the slides previously. If we work at medium or relatively high concentration, relative quantities are better than absolute and these concentrations roughly around 1 000 µg/kg are quite high in terms of acrylamide determination. So that we now need to calculate the relative u(Rw) and this is done by dividing the absolute u(Rw) by the average value. And the average value of course?? Cause! is this one here. And also, in order to present this value in percentages, it is useful to multiply by 100. So it's 2,7 % relative. And the calculations that I just performed in Microsoft Excel are also presented here in this slide. Let us look now how to find the bias component of measurement uncertainty and again I will start with Microsoft Excel. For bias component, we need to do more calculations than for the reproducibility component. First of all, we have two certified reference materials. So we need two reference values here. And we go and get these reference values from the data sheet. So, secondly we need the measurement uncertainties of those reference values, and the expanded uncertainties at k2 level are also given in this sheet. Most of the time the measurement uncertainties of the certified values of certified reference materials come as k2 uncertainty. However, for calculating our measurement uncertainty we will need standard uncertainties. Therefore, both of these we will need to divide by 2. So that now we have the standard uncertainties or, to be more exact, combined standard uncertainties of the reference values, which we can also call as in fact u (Cref). Now the laboratory result here, we have to use the mean values obtained at our laboratory. And the mean value, we get from here. So that here we now have all the data in absolute terms. Now, this Uc we also need to calculate as relative because afterwards we will use it in relative terms. And this is done very easily. We divide the uncertainty by the value and multiply by 100. Just the same is done also. So, now we can find the bias and the bias is found as laboratory result minus the reference value. And also here, in the laboratory result minus the reference value. And these bias values also now are absolute values not relative values, but we need to calculate relative values. And for calculating relative bias values, we divide these absolute values by the reference values and multiply by 100. So, now we have calculated 2 bias estimates and now we can start combining them. And combining goes via the roadmap that we have looked at, and let us take a brief look at it again. So we will first average the bias values themselves using this RMS bias formula and secondly we will also average the u(C ref) values as seen here. So we do 2 mean squares averaging operations now. So let us start by RMS bias is equal to sqrt (sum of squares of the relative bias values divided by the number of bias estimate. And the number of bias estimates in our case is 2. So that we can find very easily and it will also be in percent, as it was the case with the bias values themselves. And now, u (C ref) relativ, we find it in a similar way: square root of sum of squares of these relative values. and again we divide by 2 because we have t2 estimates. And eventually we calculate now the u( bias) relative according to this formula here. Square root of sum of squares of these values, and again in percentages. The bias data that we just calculated in Excel are now presented here in this slide. And if we look at these data we see that u(bias) which is composed of two component: the uncertainty of the reference values and the RMS bias. In this case has those components at rather similar levels. So they contribute quite similarly to the bias uncertainty. It's in fact more usual that the RMS bias component is larger than the u(C ref) but here the difference is quite small. This means the laboratory has done quite good job in determining acrylamide of it in these reference materials. Now, since we have both the reproducibility and the bias component of uncertainty calculate, we can now calculate the combined standard uncertainty and then the expanded uncertainty of the result. And again, I will first show this in Microsoft Excel. So, we now have the u(Rw) component relative and we now have the u(bias) component relative. And we are going to combine them using this formula here. So this is our formula for calculating the combined standard uncertainties. And first this combined standard uncertainty will be calculated in relative terms because all our calculations were done in relative terms. And again sqrt, square root sum of squares of the bias value and the relative reproducibility. And this of course will be in percentage, this uncertainty. Now if we want to assign this measurement uncertainty to this measurement value, and let's remember: in the Nordtest approach we always assign the uncertainties that we calculate to the measured values, then all we have to do is to multiply this uncertainty given in percentage by this value here and then divide by 100, let's do that. Multiplied by this value and divided by 100. So 48. And this value now already is in µg/kg. So. And the expanded uncertainty we can now find easily by multiplying the combined standard uncertainty with the coverage factor which in our case is 2. Again this 9.5 is the relative expanded uncertainty. And now the absolute expanded uncertainty, we can calculate by multiplying the relative combined standard uncertainty by the same coverage factor: 2. And of course the unit here will again be µg/kg. So we have now managed to find the uncertainty for our result. This slide now summarized is our measurement uncertainty calculation. We have here, the same uncertainties as they were calculated in Microsoft Excel, presented in different ways. And finally the result presentation is here: expanded uncertainty, value, unit, the recoverage factor and also I have written here norm. It is now we're thinking: can we assume normal distribution here or not? It turns out that if we look at the components of the uncertainty, the way they are found here, we discover that in broad terms we again have three components which are roughly comparable by the magnitude. Let's see, we see that u(C ref) rel and RMS bias rel, they are both very similar as I already mentioned previously. But also not too different is the relative reproducibility term. So that since we have three roughly similarly important uncertainty sources, we can assume the normal distribution. In fact, when we do measurement uncertainty calculation by the Nordtest approach, then even if those components are not so similar, which often happens, then usually still we can assume normal distribution because in fact, all of these components inturn come from different kinds of data. So that eventually, we actually have many different influences and uncertainty sources taking into account, because long time periods are used also several reference materials which all have uncertainties and all laboratory determinations have uncertainties. So combining all these effects together usually leads to a sufficiently large number of different effects to enable them as saying that the distribution is roughly normal. So that in the case of the Nordtest measurement uncertainty calculation, if there are sufficient data available, then usually normal distribution can be assumed. | Regardons maintenant comment l'approche de validation par un laboratoire unique pour l'approche Nordtest fonctionne en pratique. Et nous étudions cela sur l'exemple de la quantification de l'acrylamide dans des en-cas par LC-MS. L'acrylamide est un composé toxique qui peut, parfois, être présent dans la nourriture produite par processus chauffant par exemple les chips, le pain croustillant, etc… Puisque c'est un composé toxique et que sa teneur, dans les en-cas, peut atteindre des niveaux assez élevés, la quantification de l'acrylamide dans de tels aliments est très importante. Comme je l'ai dit précédemment dans l’approche de validation par un laboratoire unique, l'incertitude est plutôt calculée sur la procédure de la méthode analytique que sur le résultat en particulier. Et ce que nous allons maintenant faire, c'est calculer l'incertitude sur la procédure et ensuite l'appliquer au résultat. Le laboratoire a trouvé une teneur de 998 µg/kg et c'est notre résultat pour lequel nous allons, finalement, assigner l'incertitude. Maintenant, pour trouver le biais ainsi que la composante de reproductibilité de l'incertitude, le laboratoire a deux matériaux de référence certifiés. L'un d'entre eux est un paquet de chips et l'autre du pain croustillant. Pour ces deux aliments, il y a des valeurs de références disponibles et dans le cas du pain croustillant, ce dernier a également été utilisé comme un échantillon de référence par le laboratoire. Cela veut dire que le laboratoire possède une quantité suffisante d'échantillon et qu'il a été analysé de nombreuses fois sur une longue période. Regardons maintenant quelles sont les données de ces CRM (Matériaux de Référence Certifiés) et nous pouvons les voir ici. Ce sont les valeurs de référence de l'acrylamide. Et ce sont les incertitudes élargies à k=2. Regardons maintenant les données que le laboratoire a pour ces matériaux de référence certifiés. Avec le pain croustillant, un certain nombre de quantification a été réalisé sur différents jours et toutes ces valeur réunies conduisent à la valeur moyenne du laboratoire qui peut-être comparée à la valeur de référence du certificat et donc être utilisée pour la détermination du biais. D'autre part, puisque la quantification a été réalisée sur une longue période de temps, plus d'une année, les valeurs peuvent aussi être utilisées pour déterminer la reproductibilité intra- laboratoire à longue terme ou la composante tenant compte des effets aléatoires. Ce matériau de référence certifié peut donc être utilisé simultanément pour ces deux objectifs. L'autre matériau de référence certifié a également été analysé plusieurs fois, ce qui mène, à nouveau, à cette valeur moyenne. Cela signifie que nous pouvons utiliser cette valeur pour la détermination du biais. Mais, pour la reproductibilité intra- laboratoire à long terme, nous ne pouvons pas utiliser ces résultats car toutes les quantifications ont été réalisées durant une période de temps relativement courte et sur deux jours uniquement. Notre évaluation de l'incertitude sera effectuée selon la feuille de route que nous avons déjà vu précédemment et qui est donnée ici. Donc, de l'analyse d'un MRC, nous pouvons obtenir cette incertitude due aux effets aléatoires, c'est la reproductibilité intra-laboratoire. Et deuxièmement, de ces deux CRM, nous pouvons obtenir la valeur de biais. Ces deux CRM nous donnent donc une seule valeur de biais, que nous moyennons. Ces deux CRM possèdent également une valeur u(C ref) chacun, ce qui correspond à une incertitude sur chacune des valeurs de référence, incertitudes que nous moyennons également, puis nous calculons le u(biais). Enfin, u(Rw) et u(bias) sont combinés ensemble pour former l'estimation de l'incertitude-type composée. Pour savoir plus précisément comment ces calculs sont réalisés, nous allons étudier un exemple concret avec de vraies données en utilisant Microsoft Excel. J'ai préparé une feuille de calcul où j'ai déjà rentré plusieurs paramètres dont nous aurons besoin pour les calculs. Et également, j'ai mis, ici, la valeur du résultat de la mesure. De plus, nous avons ici la feuille où toutes les données sont fournies: à la fois sur les valeurs de références et les incertitudes associées aux deux matériaux de référence, mais aussi toutes les mesures en parallèles et les valeurs que le laboratoire a déterminées. Et, nous allons maintenant regarder comment les données que nous avons ici, sur cette feuille, seront utilisées pour calculer l'incertitude de mesure dans cette feuille là. Nous allons commencer avec la composante de reproductibilité. Comme je l'ai dit, nous pouvons seulement utiliser le premier matériau de référence certifié pour l'évaluation de la reproductibilité. Et celle-ci, sera évaluée simplement comme étant l'écart-type de ces mesures intra-laboratoire (réalisées sur une longue période). Donc ce u(Rw) est en fait égal à l'écart-type des mesures faites au laboratoire. Et, bien sur, nous devons indiquer les unités. Donc ce sont des µg/kg. Maintenant, dans ce cas, nous allons utiliser des grandeurs relatives pour le calcul d'incertitudes comme nous l'avons vu dans une des diapos précédentes. Si nous travaillons à moyenne ou relativement hautes teneurs, alors des grandeurs relatives sont plus adaptées que les grandeurs absolues et, ces teneurs aux alentours de 1 000 µg/kg, sont assez élevés en terme de teneur en d'acrylamide. Nous avons maintenant besoin, de calculer le u(Rw) relatif et ceci est réalisé en divisant le u(Rw) absolu par la valeur moyenne. Et la valeur moyenne, bien sur, est celle-ci ici. De plus, dans le but de présenter ce résultat en pourcentage, il est utile de multiplier par 100. Donc ça fait 2,7 % en relatif. Et les calculs que je viens juste de réaliser dans Microsoft Excel sont également présentés, ici, dans cette diapositive. Regardons maintenant comment trouver la composante de biais de l'incertitude de mesure et, à nouveau, je vais démarrer avec Microsoft Excel. Pour la composante de biais, nous avons besoin de faire plus de calculs que pour la composante de reproductibilité. Tout d'abord, nous avons deux matériaux de référence certifiés. Donc nous avons besoin de deux valeurs de référence. Et nous allons obtenir ces valeurs de référence à partir de cette feuille de calcul là. Ensuite, nous avons besoin des incertitudes de mesures de ces valeurs de référence là et les incertitudes élargies à k=2 sont également données dans cette feuille ci. La plupart du temps, les incertitudes de mesures des valeurs de référence des matériaux de référence certifiés sont indiquées pour k=2. Cependant, pour calculer notre incertitude de mesure, nous aurons besoin des incertitudes-types. Par conséquent, ces deux incertitudes devront être divisées par 2. Donc maintenant, nous avons les incertitudes-types ou, pour être plus exact, les incertitudes-types composées des valeurs de référence qui peuvent, en fait, être aussi notée u(Cref). Maintenant, pour la case résultat de laboratoire, nous devons utiliser la valeur moyenne obtenue pour notre laboratoire. Et la valeur moyenne, nous l'obtenons à partir d'ici. Donc maintenant nous avons toutes les données en termes absolus. Maintenant ces Uc ici, nous devons aussi les calculer en relatif parce qu'ensuite, nous les utiliserons comme termes relatifs. Et cela se fait assez facilement. Nous divisons l'incertitude par la valeur de référence et nous multiplions par 100. La même chose est également réalisée ici. Maintenant, nous pouvons trouver le biais et ce dernier est obtenu à partir du résultat de laboratoire - la valeur de référence. De même, ici, résultat du laboratoire - valeur de référence. Et ces valeurs de biais sont maintenant aussi des valeurs absolues et non des valeurs relatives, or, nous avons besoin de calculer des valeurs relatives. Et, pour calculer des valeur de biais relatives, nous divisons ces valeurs absolues par les valeurs de références et nous multiplions par 100. Maintenant, nous avons calculé deux estimations de biais et nous pouvons commencer à les combiner. Et leur association se fait à l'aide de la feuille de route que nous avons observé, et jetons-y, à nouveau, un rapide coup d'œil. Donc, dans un premier temps nous moyennerons les valeurs de biais ensemble en utilisant la formule du RMS biais et ensuite nous moyennerons aussi les valeurs de u(Cref) comme indiqué ici. Donc, nous calculons maintenant 2 moyennes quadratiques pondérés. Démarrons avec le RMS biais qui est égal à Racine carrée (Somme des carrés des biais relatifs divisée par le nombre de biais estimé). Le nombre de biais estimé dans notre cas est à 2. Ce qui fait que nous pouvons trouver très facilement le RMS biais et il sera également en pourcentage, comme c'était le cas avec les valeurs de biais. Maintenant u(C ref) relatif , nous pouvons le trouver de manière similaire: Racine carrée (Somme des carrés de ces valeurs relatives) et à nouveau nous divisons par 2 car nous avons deux estimations. Finalement, nous calculons le u(biais) relatif selon cette formule ici : Racine carré (Somme des carrés de ces valeurs) et à nouveau, en pourcentage. Les valeurs de biais que nous venons juste de calculer dans Excel sont maintenant présentées sur cette diapo. Et si nous regardons ces données, nous voyons que u(biais) est composé de deux morceaux d'incertitude: l'incertitude sur les valeurs de référence et le RMS biais. Ici, ces composantes ont un poids similaire. Donc ils contribuent de manière assez similaire à l'incertitude du biais. En fait, il est plutôt habituel que la composante du RMS biais contribue davantage que le u(C ref) mais ici la différence entre ces 2 composantes est plutôt faible. Cela veut dire que le laboratoire a bien travaillé lors de la quantification de l'acrylamide dans ces matériaux de référence. Maintenant, puisque que nous avons calculé à la fois l'incertitude de la composante de reproductibilité et de biais, nous pouvons maintenant calculer l' incertitude-type composée et ensuite l'incertitude élargie du résultat. Et à nouveau, je vais montrer ceci dans Microsoft Excel. Donc nous avons maintenant la composante u(Rw) relative ainsi que la composante u(biais) relative. Et nous allons les combiner en utilisant la formule juste là. Voici notre formule pour calculer les incertitudes-types composées. Et, dans un premier temps, ces incertitudes-types composées seront calculées en valeurs relatives car tous nos calculs ont été faits en valeurs relatives. A nouveau Racine carrée (Somme des carrés des biais et des reproductibilités relatives). Et, bien sur, ceci sera en pourcentage, cette incertitude. Maintenant, si nous voulons assigner cette incertitude de mesure à ce résultat de mesure et souvenons nous : dans l'approche Nordtest, nous attribuons toujours l'incertitude que nous calculons aux valeurs mesurées. Alors tout ce que nous avons à faire, c'est de multiplier cette incertitude donnée en pourcentage par cette valeur ici et ensuite diviser par 100, faisons le. Multipliée par cette valeur et divisée par 100. Ce qui fait 48. Cette valeur est déjà en µg/kg. Et l'incertitude élargie, nous pouvons la trouver facilement en multipliant l'incertitude-type composée par le facteur d'élargissement qui, dans notre cas, vaut 2. A nouveau ce 9,5 est une incertitude élargie relative. Maintenant l'incertitude élargie absolue, nous pouvons la calculer en multipliant l'incertitude-type composée relative par le même facteur d'élargissement: 2. Et bien sur l'unité ici est encore des µg/kg. Nous avons maintenant réussi à trouver l'incertitude pour notre résultat. Cette diapo récapitule nos calculs d'incertitude de mesures. Nous retrouvons, ici, les mêmes incertitudes qui ont été calculées dans Microsoft Excel, présentées de manière différente. Et finalement la présentation du résultat est ici: incertitude élargie, valeur, unité, facteur de recouvrement et aussi j'ai écrit, ici, norm. Réfléchissons maintenant: pouvons-nous présumer une distribution normale dans ce cas là ou non? Il s'avère que si nous regardons les composantes de l'incertitude, la manière dont elles ont été trouvées, ici, nous remarquons, qu'en manière générale, nous avons à nouveau 3 composantes qui sont plus ou moins semblables de part leurs valeurs. Observons, nous constatons que u(C ref) rel et RMS biais rel sont tout les deux très similaires comme je l'ai déjà évoqué précédemment. De plus, la reproductibilité relative n'est pas non plus très différente de ces deux termes. Donc puisque nous avons approximativement trois importantes sources d'incertitude similaires, nous pouvons supposer que la distribution est normale. En fait, quand on fait des calculs d'incertitude de mesures par l'approche Nordtest, même si les composantes ne sont pas très semblables, ce qui arrive souvent, alors nous pouvons quand même, assez souvent, présumer une distribution normale car, en fait, toutes les composantes proviennent de différents types de données. Donc, en réalité, nous avons de nombreuses influences et sources d"incertitudes différentes prises en compte car de longues périodes de temps sont utilisées ainsi que plusieurs matériaux de référence qui ont tous des incertitudes et tous les résultats de laboratoire possèdent des incertitudes. Donc, la combinaison de tous ces effets conduit souvent à un nombre suffisamment important d'effets différents, ce qui nous permet de dire que la distribution est à peu près normale. Donc dans le cas des calculs d'incertitude de mesure de l'approche Nordtest, s'il y a suffisamment de données disponibles, alors souvent la distribution normale peut être supposée. |
Quelques commentaires sur cet exemple :
1- Nous utilisons des incertitudes relatives dans cet exemple. La raison en est que les teneurs dans ces échantillons sont assez élevées et en analyse par LC-MS, la variabilité des résultats est souvent plus ou moins proportionnelle à la valeur de ces résultats.
2- Dans cet exemple, nous présumons que les matrices des CRMs utilisées, le paquet de chips et le pain croustillant, sont suffisamment similaires. Cela veut donc dire que la reproductibilité et le biais obtenus avec ces matrices sont également semblables. Dans ce cas, l’incertitude calculée est valable pour les deux matrices. Si, par la suite, nous analysons des matrices différentes de celles des chips et du pain croustillant alors nous ne pourrons pas appliquer, pour ce résultat-là l’estimation d’incertitude obtenue avec les chips et le pain croustillant. Pour évaluer si une nouvelle matrice est suffisamment proche de celle utilisée pour l’évaluation du biais, il faut généralement se baser sur l’expérience pratique du manipulateur.
3- L’incertitude estimée Ucrel que nous avons obtenu est une incertitude relative, donc elle est associée à un résultat CA par la formule suivante :
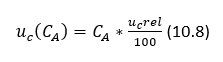
En principe, cette incertitude peut être assignée à un résultat de n’importe quelle valeur, mais, en pratique, il ne serait pas correct d’appliquer cette incertitude à des résultats analytiques qui sont très différents de ceux présentés ici. En termes de différence, il ne devrait pas y avoir plus qu’un facteur 3 voir 4 entre les résultats.
Quand nous utilisons l’approche Nordtest pour l’estimation d’incertitude, alors, en général, nous pouvons supposer que le nombre de degrés de liberté est suffisamment grand pour que l’incertitude élargie (k=2) soit supposée représenter un intervalle de confiance au seuil de 95 % de probabilité.
***
[1] Veuillez noter que dans les tableaux de la diapositive « Measurements with the CRMs » les unités de mesures doivent être en µg/kg et non en mg/l.
Les diapositives de cet exemple et les fichiers de calcul (initial et résolu) sont disponibles ci-dessous :
nordtest_uncertainty_example_acrylamide_lc-ms.pdf
12. Comparaison des approches
Résumé : Cette partie résume les principales caractéristiques des méthodes d'estimation de l'incertitude, leurs avantages et leurs inconvénients.
Dans la mesure du possible, il convient d'utiliser l’une des approches dites "de laboratoire unique". Les approches inter-laboratoires ne conviennent que pour obtenir des estimations d'incertitude très grossières. Nous recommandons de ne les utiliser que si le laboratoire ne dispose pas encore de la méthode de mesure et souhaite savoir, approximativement, quelle incertitude peut être obtenue.
Si le laboratoire possède les compétences et le temps nécessaires pour mener les investigations sur les procédures analytiques, l'approche par modélisation est souvent appropriée. Si le laboratoire dispose d'un temps limité, mais qu'il possède déjà des données de validation et de contrôle qualité, alors l'approche de validation par un laboratoire unique est la plus appropriée.
Comparaison des approches d'estimation d'incertitudes de mesures
http://www.uttv.ee/naita?id=17917
| Let us now compare the uncertainty estimation approaches that we have seen. They are all combined with their pros and cons properties into this slide. So if we start by the modeling approach and as you've probably noticed it involves quite extensive calculations and it's not very easy to apply, so it gives good results mostly with quite advanced laboratories because quite some knowledge is required. The knowledged and extra work is also usually required because specific experiments have to be carried out often times to evaluate the uncertainty components. And if some uncertainty component is left out of consideration and if it is an important component then there is always the danger to underestimate the uncertainty. The same goes if some component is taken into consideration but it is underestimated but on the other hand it gives a lot of information about your procedure and thereby promotes thinking about it. And also it has high value in teaching. It is very good for students to see where the uncertainty actually comes from. By this, this modeling approach stands out from all the others. Now if we look at the single-lab validation approach then this should be fairly well suitable for routine laboratory who do not have time or competence for carrying out lot of extra studies. Lots of the data are needed but large amount of that data comes out automatically. So laboratories have to do validations of their procedures anyhow. They usually have to analyze certified reference materials anyhow. And they have to participate in inter-laboratory comparisons anyhow. And all this data can directly be used for the measurement of uncertainty evaluation So that only known less extra work is required. The data that automatically are available in the laboratory can be used directly. And the uncertainty estimates that are obtained are very realistic so the danger to underestimate measurement uncertainty is much lower. In fact, if the bias estimate is obtained from not very trustworthy sources or if the reference values are not good enough this approach can give you overestimated uncertainty. But of course unfortunately the teaching value of this approach is lower than of the modeling approach and it does not give much information about the procedure. And finally the inter laboratory approaches, inter laboratory validation proficiency testing approach which i only briefly mentioned and for which I said that i do not really recommend them, these approaches in fact do not need any knowledge and also no data from your own laboratory. But the uncertainties that they give are so crude that they can be used only as first estimates. And I would recommend to use them only in the case if you do not yet have the procedure set up in your laboratory and you would simply like to see what uncertainty you could expect. In all other cases, when you already have the procedure in your lab and you have a data and you work with it, I strongly recommend not to use these but to use either of these two approaches. | Comparons maintenant les méthodes d'estimation d'incertitude que nous avons vues. Elles sont toutes regroupées avec leurs avantages et inconvénients dans cette diapo. Si nous commençons par la méthode de modélisation et comme vous l'avez probablement remarqué, elle implique des calcules assez compliqués et elle n'est pas très facile à appliquer, donc elle permet d'avoir de bons résultats pour des laboratoires assez avancés en général car elle nécessite certaines connaissances assez avancées. Elle nécessite aussi en général des connaissances et du travail en plus car souvent, des expériences très spécifiques doivent être réalisées pour évaluer les éléments d'incertitude. Si un certain élément d'incertitude est laissé hors de considération et si c'est un élément important, alors il y a toujours le danger de sous-estimer l'incertitude. Il y a le même soucis si un élément est pris en considération mais il est estimé. D'un autre côté, cela permet d'avoir beaucoup d'information sur la procédure et ainsi favorise son questionnement. Cela a aussi une grande valeur dans l'enseignement. C'est très bien pour les élèves de voir la provenance des incertitudes. Par cela, cette approche de modélisation se démarque de toutes les autres. Maintenant, si nous regardons la méthode de validation de laboratoire unique alors cela devrait être assez convenable pour des laboratoires de routine qui n'ont pas le temps ou pour effectuer beaucoup d'études supplémentaires. Beaucoup de données sont nécessaires mais une grande quantité de ces données est extraite automatiquement. Donc les laboratoires doivent faire des validations de leurs procédures. Mais de toute façon, ils doivent en général analyser des matériaux de référence certifiés et il doivent participer à des comparaisons inter-laboratoires. Et toutes ces données peuvent être directement utilisées pour les mesures d'évaluation d'incertitude. Pour que moins de travail soit requis. Les données qui sont automatiquement à la disposition dans le laboratoire peuvent être utilisées directement. Et les estimations d'incertitude qui sont obtenues sont très réalistes donc le danger de sous-estimer les incertitudes de mesure est moindre. En effet, si l'estimation biaisée est obtenue de sources pas très fiables ou que les valeurs de référence ne sont pas assez bonnes, cette méthode peut donner des incertitudes sur-estimées. Mais bien sûr malheureusement, la valeur instructive de cette approche est moindre que celle pour la méthode de modélisation et elle ne donne pas beaucoup d'informations sur la procédure. Et finalement, les approches inter laboratoires, l'approche de test de compétence de validation inter laboratoire que j'ai seulement brièvement mentionné et pour lequel j'ai dit que je ne les recommandais pas trop, ces approches ne requièrent en fait aucune connaissance mais aussi aucune donnée du laboratoire propre. Mais les incertitudes qu'ils donnent sont tellement brutes qu'elles ne peuvent être utilisées seulement comme une première estimation. Et je recommanderais de seulement les utiliser dans le cas où la procédure n'a pas encore été mise en place dans le laboratoire et où l'on veut simplement voir quelle incertitude on peut s'attendre à avoir. Dans tous les autres cas, quand la procédure est déjà mise en place dans le laboratoire et on a des données et on peut travailler avec, je recommande fortement de ne pas utiliser ces approches mais d'utiliser une des deux autres méthodes. |
Le tableau 11.1 résume les avantages et les inconvénients de ces approches. Voir également le tableau 8.1 de la partie 8 pour certaines différences essentielles.
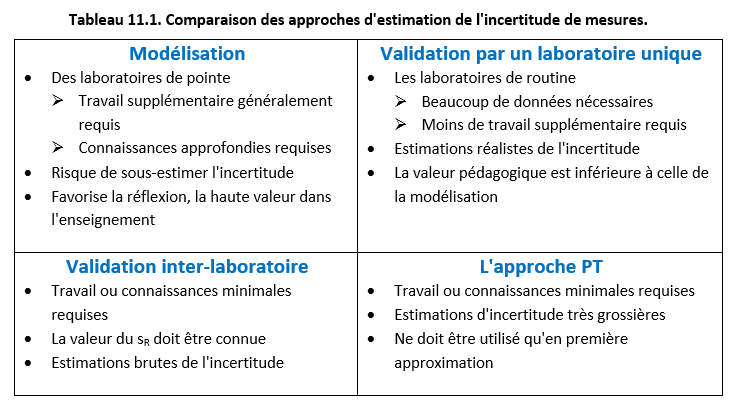
Autoévaluation sur cette partie du cours : Test 11
13. Comparaison de résultats de mesure
https://sisu.ut.ee/measurement/12-using-measurement-uncertainty-estimates-decision-making
Résumé : Cette section explique que les estimations de l'incertitude de mesure sont indispensables si l'on veut comparer deux résultats de mesure.
Comparaison de résultats de mesure
http://www.uttv.ee/naita?id=18095
| It very often happens that we have 2 different measurement results for the same quantity and we would like to know do they agree or are they in disagreement. How do we know that ? There are several approaches how to establish whether two measurement results are in agreement or not that some of them quite sophisticated but we will look here at a very simple approach which is based on the so called Zeta score. The Zeta score of two measurement results is found as follows. It's the ratio of the difference between result 1 and the result 2 and it is divided by the square root of sum of squared combined standard uncertainties Let's see how this works in reality. Suppose we have two different measurement results. X1 is equal to 4,5 milligrams per kilogram and X2 is equal to 3,5 milligrams per kilogram. Seemingly they are very different but if these are let's say some pesticide determination or other kind of trace analysis results they usually have quite a high uncertainty and let's see what happens if they have some quite realistic uncertainties. U(x1) is equal to 0,7 milligrams per kilogram and U(X2) is equal to 0,6 milligrams per kilogram If we now calculate the z-score we will find the following, and if we make this simple calculation we find that the Zeta score is 1,1. How do we know now whether these results are in agreement or not ? The Zeta scores are evaluated according to the following rules. If the absolute value of the Zeta is below - or equal to 2 then we can say that the results are in agreement. If the Zeta values are between 2 and 3 we say that it's a warning situation. We cannot yet say that there is really disagreement but indeed the difference is still quite high. And finally, if the absolute value of the Zeta score is above 3, then we can say that yes these results are not in agreement. So based on these rules and our data we can say that even though these two results initially look rather different if we take into consideration their uncertainties they actually agree very well because 1,1 certainly smaller than 2 ! | Il arrive très souvent que nous ayons 2 résultats de mesure différents pour la même quantité et nous voudrions savoir s'ils sont en accord ou s'ils sont en désaccord. Comment le savons-nous ? Il y a différentes approches pour établir si deux résultats de mesure sont en accord ou pas dont certaines sont assez sophistiquées mais nous allons voir ici une approche très simple qui est basée sur ce qu'on appelle le score zêta. Le score zêta de deux résultats de mesure s'écrit comme suit. C'est le ratio de la différence entre le résultat 1 et le résultat 2 divisé par la racine carrée de la somme des incertitudes type composées. Maintenant voyons comment ça fonctionne en réalité. Supposons que nous ayons 2 résultats de mesure différents. X1 est égal à 4,5 mg/kg et X2 est égal à 3,5 mg/kg. De toute évidence ils sont très différents mais s'il s'agit de déterminer des pesticides ou d'autres sortes de résultats d'analyses de traces les résultats ont en général plutôt une grande incertitude. Voyons donc ce qu'il se passe s'ils ont des incertitudes assez réalistes : U(X1) est égale à 0,7 mg/kg et U(X2) est égale à 0,6 mg/kg. Si nous calculons maintenant le score z nous allons trouver la chose suivante. Et si nous faisons ce calcul simple on trouve que le score zêta est de 1,1. Comment savons-nous maintenant si ces résultats sont en accord ou non ? Les scores zêta sont évalués selon les règles suivantes. Si la valeur absolue de zêta est inférieure ou égale à 2 alors on peut dire que les résultats sont en accord. Si les valeurs de zêta sont entre 2 et 3 nous pouvons dire que c'est une situation critique. Nous ne pouvons pas encore dire s'ils sont vraiment en désaccord mais effectivement la différence est assez grande. Et finalement, si la valeur absolue du score zêta est supérieure à 3, alors on peut dire que oui, ces résultats ne sont pas en accord. Donc en se basant sur ces règles et nos données, nous pouvons dire que même si ces 2 résultats semblaient plutôt différents initialement, si on prend en considération leurs incertitudes ils sont en réalité tout à fait en accord car 1,1 est assurément inférieur à 2 ! |
14. Autres ressources
https://sisu.ut.ee/measurement/13-additional-materials-and-case-studies
Résumé : Cette section rassemble quelques contenus, exemples et études de cas additionnels sur les techniques spécifiques d’analyses chimiques.
- 13.1. Différentes techniques analytiques
- 13.2. Estimation de l’incertitude de mesure lors de la détermination de la teneur en oxygène dissous
- 13.3. Titrage colométrique de Karl Fischer
Contenu similaire :
“ Plus de 100 expériences intéressantes en chimie”
14.1. Différentes techniques analytiques
Un grand nombre d’exemples d’estimation d’incertitude de mesure (exemple : contribution à l’incertitude) est disponible à l’adresse suivante : http://www.ut.ee/katsekoda/GUM_examples/.
14.2. Estimation de l'incertitude de mesure lors de la détermination de la teneur en oxygène dissous
L'oxygène dissous (OD) est l'un des gaz dissous les plus importants dans l'eau. Une concentration suffisante d'OD est essentielle à la survie de la plupart des plantes et des animaux aquatiques [3] ainsi qu'au traitement des eaux usées. La concentration en OD est un paramètre clé caractérisant les eaux naturelles et les eaux usées et permettant d'évaluer l'état de l'environnement en général. Outre la concentration de CO2 dissous , la concentration en OD est un paramètre important qui façonne notre climat. Il est de plus en plus évident que la concentration d'OD dans les océans diminue [4 - 7].
Des mesures précises de la concentration d'OD sont très importantes pour étudier ces processus, comprendre leur rôle et prévoir les changements climatiques.
Les capteurs électrochimiques et optiques sont les moyens les plus répandus de mesure de la concentration d'OD. Les deux sont largement utilisés, mais les effets de différentes sources d'incertitude sur les résultats sont remarquablement différents et l'estimation de l'incertitude n'est pas simple. Afin d'aider les praticiens dans ce domaine, une validation comparative complète de ces deux différents types d'analyseurs d'oxygène dissous (DO), ampérométriques et optiques, a récemment été effectuée sur la base de deux analyseurs commerciaux représentatifs de l'OD et publiée: I. Helm, G. Karina, L. Jalukse, T. Pagano, I. Leito, Environ Monit Assess 2018 , 190 : 313 (réf [1]).
Un certain nombre de caractéristiques de performance ont été évaluées, notamment la dérive, la précision intermédiaire, la précision de la compensation de température, la précision de la lecture (dans différentes conditions de mesure), la linéarité, la dépendance au débit de la lecture, la répétabilité (stabilité de la lecture) et les effets matriciels des sels dissous. Les effets de la matrice sur les lectures dans des échantillons réels ont été évalués en analysant la dépendance de la lecture sur la concentration en sel. Les analyseurs ont également été évalués dans les mesures d'OD d'un certain nombre d'eaux naturelles. Les contributions à l'incertitude des principaux paramètres d'influence ont été estimées dans différentes conditions expérimentales. Il a été constaté que les incertitudes des résultats pour les deux analyseurs sont assez similaires, mais les contributions des sources d'incertitude sont différentes. Les résultats impliquent que l'analyseur optique peut ne pas être aussi robuste qu'on le suppose généralement, cependant, il a une meilleure stabilité de lecture, une dépendance à la vitesse d'agitation plus faible, et nécessite généralement moins d'entretien. D'autre part, l'analyseur ampérométrique a une réponse plus rapide et une plage linéaire plus large. L'approche décrite dans ce travail sera utile aux praticiens effectuant des mesures d'OD pour assurer la fiabilité de leurs mesures.
La méthode de titrage Winkler est considérée comme la méthode la plus précise pour la mesure de la concentration d'OD. Une analyse minutieuse des sources d'incertitude pertinentes pour la méthode Winkler a été effectuée et les résultats sont présentés sous la forme d'un «rapport sur l'amélioration de la méthode Winkler de haute précision pour la détermination de la concentration en oxygène dissous» .
Dans ce rapport, il est décrit comment la méthode Winkler a été optimisée pour minimiser autant que possible toutes les sources d'incertitude. Les améliorations les plus importantes ont été: la mesure gravimétrique de toutes les solutions, le prétitrage pour minimiser l'effet de la volatilisation de l'iode, la détection précise du point final ampérométrique et la prise en compte minutieuse de l'oxygène dissous dans les réactifs. En conséquence, la méthode développée est probablement la méthode la plus précise de détermination de l'oxygène dissous disponible. En fonction des conditions de mesure et de la concentration en oxygène dissous, les incertitudes-types composées de la méthode se situent dans la plage de 0,012 à 0,018 mg.dm -3 correspondant à l'incertitude élargie k=2 dans la plage de 0,023 à 0,035 mg.dm -3 (0,27 - 0,38%, relatif). Ce développement permet un étalonnage plus précis des capteurs électrochimiques et optiques d'oxygène dissous pour l'analyse de routine que ce qui était possible auparavant. La plupart de ce rapport est basé sur l'article I. Helm, L. Jalukse, I. Leito, Anal. Chim. Acta. 2012 , 741 : 21-31 (réf [2]).
Le contenu de ce cours en ligne peut être utilisé comme base pour effectuer l'évaluation de l'incertitude de mesure décrite dans le rapport susmentionné. En particulier, le Test 9.2A et le Test 9.2B sont directement liés à la mesure de la concentration d'OD.
La préparation du rapport susmentionné a été soutenue par le programme européen de recherche en métrologie (EMRP), projet ENV05 "Métrologie pour la salinité et l'acidité des océans".
- Helm, G.Karina, L.Jalukse, T.Pagano, I.Leito, Environ Monit Assess 2018 , 190 : 313.
- I. Helm, L. Jalukse, I. Leito, Anal. Chim. Acta. 2012 , 741 : 21-31.
- RF Keeling, HE Garcia, PNAS, 2002 , 99 (12): 7848 - 7853.
- G. Shaffer, SM Olsen, JOP Pedersen, Nat. Geosci. 2009 , 2 : 105-109.
- RF Keeling, A. Körtzinger, N. Gruber, Annu. Rév. Mar. Sci. 2010 , 2 : 199-229.
- D. Gilbert, NN Rabalais, RJ Diaz, J. Zhang, Biogeosci. Discuter. 2009 , 6 : 9127–9160.
- JPA Hobbs, CA McDonald, Journal Fish Biol. 2010 , 77 : 1219-1229.
14.3. Titrage coulométrique Karl Fischer
Les ingrédients actifs des produits pharmaceutiques, les composites à base de fibres de carbone, les polymères, les nouveaux papiers actifs à base de cellulose, les poudres alimentaires, la biomasse - tous ces produits et de nombreux autres matériaux solides sont fortement affectés par l'humidité lors de leur transformation en divers produits. Les erreurs et les inconsistances dans la mesure et le contrôle de l'humidité dans les processus industriels entraînent une diminution de la vitesse/du débit des processus et une augmentation du gaspillage, une réduction de la durabilité des biomatériaux, une augmentation de la consommation d'énergie pour le séchage et une augmentation des émissions de particules fines lors de la combustion de la biomasse.
La méthode coulométrique Karl Fischer est actuellement la méthode la plus précise de mesure de l'humidité. L’étude des facteurs déterminant l'incertitude de la méthode coulométrique de titrage Karl Fischer a été réalisée.
Cette étude donne un aperçu des facteurs qui déterminent l'incertitude de la méthode coulométrique Karl Fischer (cKF) pour la détermination de la teneur en eau. Une distinction est faite entre les sources d'incertitude provenant de la méthode cKF elle-même et les sources d'incertitude dues à la manipulation de l'échantillon. Les sources d'incertitude "composées" - répétabilité, reproductibilité et biais qui incorporent en fait les contributions de ces deux catégories de sources d'incertitude - sont également brièvement abordées.
D'après les données de la littérature, les sources d'incertitude ayant le plus d’influence dans la méthode de titrage coulométrique KF sont les possibles interférences chimiques, l'instabilité de l'instrument et la précision de la détermination du point final. Les sources d'incertitude dues à la manipulation des échantillons sont plus problématiques avec les échantillons solides qu'avec les échantillons liquides. Les sources d'incertitude les plus importantes liées à la préparation de l'échantillon sont la modification de la teneur en eau de l'échantillon avant la mesure et le transfert incomplet de l'eau de l'échantillon au milieu de réaction.
La conclusion générale est que, bien que les sources d'incertitude de la méthode cKF soient en général assez bien connues, la discussion est presque toujours soit qualitative, soit limitée aux sources d'incertitude composées, et il y a très peu d'informations quantitatives disponibles sur les contributions des sources d'incertitude réelles.
La préparation de ce rapport a été soutenue par le programme européen de recherche en métrologie (EMRP), projet SIB64 METefnet "Metrology for Moisture in Materials".

(4.12){kind=link}
coulometric_kf_titration_measurement_uncertainty_sources_survey.pdf
15. Autotests
https://sisu.ut.ee/measurement/13-tests-and-exercises
Cette partie rassemble tous les tests et exercices de ce cours.
- Le concept d'incertitude de mesure (MU) - Test 1
- L'origine de l'incertitude de mesure - Test 2
- La fonction normale de distribution - Test 3.1
- Moyenne, écart type et incertitude de mesure - Test 3.2
- Écart type de la moyenne - Test 3.4
- Distribution rectangulaire et triangulaire - Test 3.5
- La distribution de Student - Test 3.6
- Quantification des sources d'incertitude - Test 4.1
- Calcul de l'incertitude type composée - Test 4.2
- Focus sur l'incertitude obtenue - Test 4.3
- L’incertitude élargie - Test 4.4
- Présentation des résultats de mesure - Test 4.5
- Les sources d'incertitude de mesure - Test 5.3
- Traitement des effets aléatoires et systématiques - Test 5.4
- Retour sur les effets aléatoires et systématiques - Test 6
- Fidélité, justesse, exactitude - Test 7
- Aperçu des approches d'estimation de l'incertitude de mesure - Test 8
- Équation du modèle - Test 9.2A et Test 9.2B
- Incertitude-type des données d’entrée - Test 9.5
- L'approche de modélisation ISO GUM - Test 9.9A , Test 9.9B et Test 9.9C
- Composantes d'incertitude associées aux effets aléatoires - Test 10.2
- Composantes d'incertitude associées aux effets systématiques - Test 10.3
- Détermination de la teneur en acrylamide dans les collations par LC-MS - Test 10.5A et Test 10.5B
- Comparaison des différentes approches - Test 11